










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Computer Journal
On-line version ISSN 2313-7835
Print version ISSN 1015-7999
SACJ vol.33 n.2 Grahamstown Dec. 2021
http://dx.doi.org/10.18489/sacj.v33i2.830
RESEARCH ARTICLE
nf-rnaSeqCount: A Nextflow pipeline for obtaining raw read counts from RNA-seq data
Phelelani T. MpangaseI, II; Jacqueline FrostII, III; Mohammed TiklyIV; Michèle RamsayI, II; Scott HazelhurstI, V
ISydney Brenner Institute for Molecular Bioscience, University of the Witwatersrand, Johannesburg
IIDivision of Human Genetics, National Health Laboratory Service and School of Pathology, Faculty of Health Sciences, University ofthe Witwatersrand, Johannesburg
IIIDepartment of Oncological Sciences, Icahn School of Medicine at Mount Sinai, New York City
IVDivision of Rheumatology, School of Clinical Medicine, University of the Witwatersrand, Johannesburg
VSchool of Electrical & Information Engineering, University of the Witwatersrand, Johannesburg
ABSTRACT
The rate of raw sequence production through Next-Generation Sequencing (NGS) has been growing exponentially due to improved technology and reduced costs. This has enabled researchers to answer many biological questions through "multi-omics" data analyses. Even though such data promises new insights into how biological systems function and understanding disease mechanisms, computational analyses performed on such large datasets comes with its challenges and potential pitfalls. The aim of this study was to develop a robust portable and reproducible bioinformatic pipeline for the automation of RNA sequencing (RNA-seq) data analyses. Using Nextflow as a workflow management system and Singularity for application containerisation, the nf-rnaSeqCount pipeline was developed for mapping raw RNA-seq reads to a reference genome and quantifying abundance of identified genomic features for differential gene expression analyses. The pipeline provides a quick and efficient way to obtain a matrix of read counts that can be used with tools such as DESeq2 and edgeR for differential expression analysis. Robust and flexible bioinformatic and computational pipelines for RNA-seq data analysis, from QC to sequence alignment and comparative analyses, will reduce analysis time, and increase accuracy and reproducibility of findings to promote transcriptome research.
Categories: · Applied computing ~ Bioinformatics
Keywords: bioinformatics, pipelines, workflows, nextflow, RNA-seq, singularity, container, reproducible
1 INTRODUCTION
With the increase in the rate at which raw sequencing data is produced due to improved technologies and reduced cost of Next-Generation Sequencing (NGS), researchers in the field of bioinformatics and computational biology are able to perform "multi-omics" data analyses to answer many biological questions (Fan et al., 2014; Kluge & Friedel, 2018; Schulz et al., 2016). However, analysis of such large datasets comes with a number of challenges, especially when it comes to sharing data analysis methods with the scientific community and being able to reproduce consistent results using the same data across different computational platforms (Boettiger, 2015; Di Tommaso et al., 2017; Kurtzer et al., 2017). When performing computational analyses of NGS data, often different tools are required at each processing step of the analysis. For example, Haas et al. (2013) describe a procedure for assembling RNA sequencing (RNA-seq) data, quantifying expression levels for transcripts and identifying differentially expressed transcripts between samples. This protocol requires a number of applications in order to be executed successfully, including Trinity, bowtie, samtools, R and NCBI-Blast + (Haas et al., 2013).
To a bioinformaticist, computational biologist or someone who is familiar with the Unix environment, installing these applications and running this protocol described by Haas et al. (2013) would be a straight-forward procedure. However, to a novice, this would be a difficult task. Not being an administrator also significantly complicates installation of applications. Another challenge in performing such a procedure would be having to re-do the analysis, either multiple times whilst changing certain parameters, or performing the analysis using more than one dataset. In this case, simply executing the protocol commands on a command line interface (CLI) would not suffice. Custom scripts would have to be created in order to compile and order the multiple commands needed to execute the protocol procedure repeatedly or on multiple datasets (Piccolo & Frampton, 2016). Another option would be to implement "workflow management systems" to construct a pipeline (or "workflow") of the multiple analyses steps, handle input/output files between applications and also automate the analysis (Di Tommaso et al., 2017; Piccolo & Frampton, 2016; Schulz et al., 2016).
Another challenge that the scientific community faces in performing multi-step analysis that requires different pieces of software at each analysis step is software dependencies and libraries (Kurtzer et al., 2017). Many bioinformatics tools are built from sources, and thus there will be a complexity of dependencies and libraries between the softwares needed to perform the analyses (Schulz et al., 2016). In addition to software and library dependency, there is also a computational environment or an operating system (OS) dependency. Installation of different application softwares on different OSs requires different configuration steps, and some applications are only designed to be executed on a specific environment of a specific OS (Kurtzer et al., 2017; Piccolo & Frampton, 2016). A solution to software and OS dependency is to use virtual machines or software package managers (containers) (Boettiger, 2015; Kurtzer et al., 2017; Piccolo & Frampton, 2016; Schulz et al., 2016).
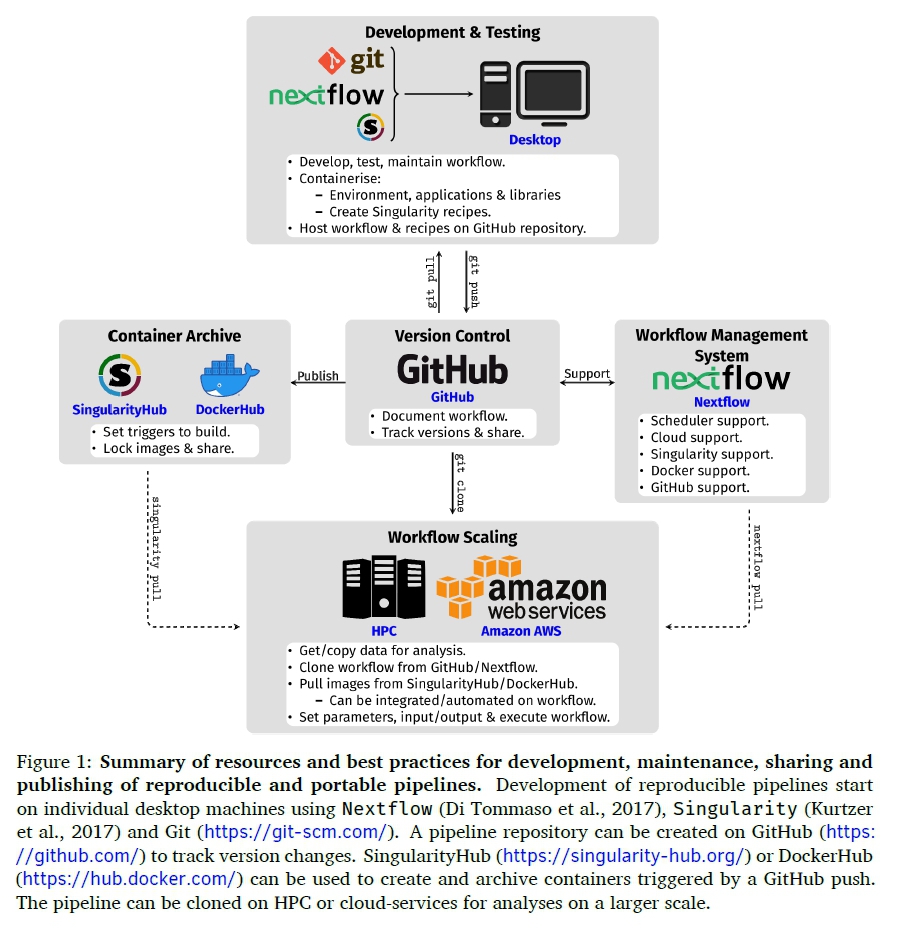
When big datasets are being analysed, personal desktop machines and laptops are not an option. In most cases, bioinformatic analyses will require a significant amount of computing power, memory and will sometimes need to be processed simultaneously (in parallel) in order to reduce the amount of time needed to perform each task (Di Tommaso et al., 2017; Kurtzer et al., 2017). These analyses have to be performed on high-performance computing (HPC) clusters available in most research institutes or cloud-computing services which offer significantly high computing resources that can meet the requirements of intensive bioinformatic analyses (Kurtzer et al., 2017). This "scaling up" of bioinformatic analyses to cloud environments and HPC clusters is further enhanced by a combination of workflow management system and con-tainerisation of software; making bioinformatic analyses pipelines "portable" across different computing platforms (Boettiger, 2015; Kurtzer et al., 2017; Piccolo & Frampton, 2016; Schulz et al., 2016). Figure 1 summarises the best practices and tools that researchers could apply to their research approach and reproducible pipeline development. This combination also overcomes the limitation of software installation on HPC clusters and cloud-services as sometimes the users do not have privileges to install softwares and their dependencies (Kurtzer et al., 2017). Furthermore, coupling the combination workflow management systems, software containers and HPC with proper documentation and storing code using version control systems (VCS) creates portable pipelines that can be shared amongst the scientific community and ensures reproducibility across different platforms (Di Tommaso et al., 2017; Kurtzer et al., 2017; Perkel, 2016; Piccolo & Frampton, 2016).
The availability of RNA-seq data from black South African patients affected with systemic sclerosis (SSc) and healthy individuals from the study by Frost et al. (2019) presented an opportunity to develop a robust computational pipeline in an effort to bridge the gap between repetitive (and most often complicated) bioinformatic analyses and the large datasets produced by NGS technologies. Human genome sequencing is still a relatively costly venture, mainly due to the genome size. However, since only a fraction of a genome is transcribed, the set of the transcribed RNA molecules (transcriptome) reflects the current state of the cell (or group of cells) in a given tissue and at a specific time. The analysis of the transcribed RNA molecules can often provide insights into the etiology and underlying pathological mechanism of a disease (Finotello & Di Camillo, 2015).
Although a useful approach, studying gene expression through the transcriptome alone has the limitation that it does not necessarily correlate directly with amount of protein present in a cell, and should therefore be interpreted with caution. Nonetheless, RNA-seq provides a quick and cost effective way of obtaining large amounts of transcriptome data, providing insights into the levels of gene expression. Such data allow us to identify transcribed genes, discover new disease-associated genes, measure transcript and gene expression abundance, study allelic information and identify splice variants for genes (Grabherr et al., 2011; Haas et al., 2013; Li et al., 2014; Trapnell et al., 2013; Trapnell et al., 2012). A typical RNA-seq analysis involves three major steps: (1) identifying a set of genes and/or transcripts from the hundreds of millions of short (~ 36-125 bases) RNA-seq reads, produced from the sequencing experiment, through mapping to a reference genome/transcriptome; (2) quantifying the abundance of the genes/transcript; and (3) performing differential expression analysis (Conesa et al., 2016). This paper presents nf-rnaSeqCount, a portable, reproducible and scalable Nextflow pipeline that addresses the first two steps of RNA-seq data analyses, i.e., (1) identification of genes from RNA-seq data and (2) quantifying their abundance.
2 PIPELINE IMPLEMENTATION AND WORKFLOW
The purpose of the pipeline is to make the task of producing raw read counts for performing differential expression analysis easier, especially for other researchers with little or no knowledge of bioinformatics. The pipeline also needs to be portable and reproducible in order to allow scaling to different computational platforms when large or small datasets are being analysed.
2.1 nf-rnaSeqCount Implementation
Nextflow (Di Tommaso et al., 2017) and Singularity (Kurtzer et al., 2017) were used to implement the pipeline into a portable and reproducible pipeline. Nextflow is a Groovy-based domain-specific language (DSL) specifically designed for bioinformaticists with strong programming knowledge to solve many of the challenges of the inability to reproduce data analysis. Some of these challenges are due to computational platform variations, software and database management, complexity of pipelines, intermediate file handling and lack of good practice (Di Tommaso et al., 2017). The advantages of Nextflow as a workflow management system are that it can handle input and output as channels between processes and reduce the need of having to create intermediate directories to store intermediate results. Variables can also be declared dynamically with no need to explicitly name files, and only the output that is required can be saved to files in each analysis step.
Nextflow also has a number of features that promote pipeline reproducibility and portability. It supports Docker1 and Singularity2, the two most used containerisation softwares in the bioinformatics community; it integrates/supports the popular VCS GitHub3 for sharing of code, and version management; and it allows for scaling of computational pipelines on HPC and cloud systems by providing out of the box scheduler support for Sun Grid Engine (SGE), PBS/Torque, Platform Load Sharing Facility (LSF), Simple Linux Utility for Resource Management (SLURM), HTCondor and Amazon Web Services (AWS) (Di Tommaso et al., 2017).
To facilitate reproducibility and portability of the pipeline, Docker containers were created for each of the processes applications in the pipeline and hosted on DockerHub4 to use with Singularity when executing the pipeline. Singularity is a lightweight platform for building and running containers that is gaining popularity in the bioinformatics community, especially for performing analysis on a large scale. It uses an image format that is supported across different versions of the C library and kernels, and gives users the ability to encapsulate their pipelines, all required applications, dependencies and base OS environment into a single file that can be locked, copied, shared and archived (Kurtzer et al., 2017). Docker containers hosted on DockerHub can be downloaded and converted into Singularity image format (SIF). Such image files have standard Linux/UNIX file permission and cannot be modified (not even by the host OS), thus can be used with confidence that nothing within the image has changed. Each SIF contains the necessary software required by each process to run. This removes the need to install all the software tools used for these analyses.
2.2 nf-rnaSeqCount Workflow
The nf-rnaSeqCount pipeline depends on Nextflow and Singularity to run. These two softwares must be installed in order for the pipeline to be executed. The input for the nf-rnaSeqCount pipeline are FASTQ files (both paired- and single-ended) for the RNA-seq data to be analysed, a reference genome (in FASTA format) and an annotation file (in GTF format) for the reference genome. These can be passed as command-line arguments during pipeline execution or specified in a configuration file (e.g. main.conf in Figure 2) that can also be passed to the pipeline during execution. Other parameters of the pipeline can be specified in a similar way. Unlike most pipelines that are executed in one go, the nf-rnaSeqCount is a modular pipeline that can be executed in multiple stages (main.nf in Figure 2), allowing the users to interact with the results at each step of the pipeline.
The different modules of the pipeline can be grouped into 4 steps, and each module (specified using the -mode argument) calls the required process in the workflow. The 4 workflow steps and their modules are as follows: (1) Data Preparation: prep.Containers and prep.Indexes; (2) Quality Control: run.ReadQC and run.ReadTrimming; (3) Alignment and Quantification: run.ReadAlignment and run.ReadCounting; and (4) Mul-tiQC: run.MultiQC. The nf-rnaSeqCount pipeline can be obtained using the following command:

The help menu for the pipeline can be accessed with the following command:

2.2.1 Data Preparation
Data preparation is mandatory at the first step of the workflow. The first process in this step, run_DownloadContainers, downloads all the required workflow containers with the required software for executing the pipeline from DockerHub and converts them to Singularity format. This step is crucial as all processes in the pipeline depend on the applications that are packaged in these containers:

Once the Singularity containers have been downloaded, the run_GenerateSTARIndex and run_GenerateBowtie2Index processes will index the reference genome (for aligning the RNA-seq reads and quantifying the abundance of the identified genomic features) using STAR (Dobin et al., 2013) and Bowtie (Langmead et al., 2009), respectively. Before indexing, the location for the reference genome (FASTA), annotation file (GTF), input FASTQ files and output directory can be provided in a configuration file (as in Figure 2), and passed to the Nextflow command using the -c option. However, the files can also be passed as command-line arguments when executing the pipeline. The remainder of the pipeline assumes that all the required files are provided in a configuration file called main.conf. To index the reference genome using STAR and Bowtie, the following commands can be used:

2.2.2 Quality Control
The nf-rnaSeqCount pipeline incorporates FastQC (Andrews, 2010) and Trimmomatic (Bolger et al., 2014) for pre-processing of reads. This "quality control" (QC) step of the nf-rnaSeqCount workflow is optional; however, it is very useful to first assess the initial quality of the reads so that poor quality reads and contaminating "adapter" sequences can be removed. In this step, the run_QualityChecks process uses FastQC to assess the quality of the RNA-seq reads. The run_ReadTrimming process, which uses Trimmomatic, is then used to remove technical sequences and poor quality bases from the data. To assess the quality of the RNA-seq reads, the following command can be executed:

Once the quality of the reads has been assessed, Trimmomatic can be used to remove low quality bases and adapter sequences. The -trim option can be used to pass Trimmomatic arguments to the pipeline:

2.2.3 Alignment and Quantification
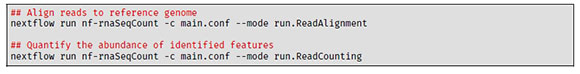
The alignment and quantification is the main step of the nf-rnaSeqCount workflow. In this step, the RNA-seq reads are aligned to the reference genome (indexed in the "Data Preparation" step) using STAR (Dobin et al., 2013) in the run_ReadAlignment process. The resulting BAM files are then used to quantify the abundance of the identified genes using both featureCounts (Liao et al., 2014) (run_FeatureCounts process) and htseq-count (Anders et al., 2015) (run_HTSeqCount process). To align the reads to the reference genome and quantify the abundance of the genomic features identified, the following commands can be used:
2.2.4 MultiQC
The MultiQC step is optional. In this step, the run_MultiQC process uses MultiQC (Ewels et al., 2016) to collect all the statistics from all the programs that were executed in the workflow, and give a summary of all statistics in an HTML file. To obtain the summary statistics of the workflow, the following command can be used:

2.3 nf-rnaSeqCount Pipeline Output
The output directory for the nf-rnaSeqCount pipeline contains a number of folders:
• n number of folders corresponding to each of the samples that were processed by the pipeline. These folders contain general statistics on mapping using STAR.
• featureCounts folder containing read counts matrix (gene_counts_final.txt) for htseq-count. This file can be used for differential expression analysis.
• htseqCounts folder containing read counts matrix (gene_counts_final.txt) for featureCounts. This file can be used for differential expression analysis.
• report_QC folder containing MultiQC QC reports in HTML format. This file can be used to assess the quality of read mapping and gene quantification.
• report_workflow folder containing pipeline execution reports. These files can be used to trace the execution of the pipeline and check other metadata in order to assign resources correctly to the processes.
2.4 Testing
The RNA-seq data of black South African patients with SSc were used to test and validate the usefulness of the nf-rnaSeqCount pipeline. This transcriptome data was from a study by Frost et al. (2019), conducted with ethics approval of the Human Research Ethics Committee (HREC [Medical]) of the University of the Witwatersrand (certificate number M120512). The nf-rnaSeqCount pipeline was initially developed and tested on the University of the Witwatersrand (Wits) Computing cluster using SLURM and PBS job schedulers. The pipeline also has been successfully tested on the University of Cape Town (UCT) eResearch HPC5 and on Amazon's AWS6 using RNA-seq data for this study. On the UCT's eResearch HPC, which has SLURM as the job scheduler, the same computing requirements as with the Wits Computing cluster were used.
For AWS execution of the pipeline, the Nextflow supported Amazon Machine Image (AMI), ami-4b7daa32, was used to deploy an Amazon Elastic Block Store (EBS) of 1000GB using the Elastic Compute Cloud (EC2), m4.10xlarge, with 40 virtual CPUs and 160 GB of memory. All AWS analyses were performed on the European (Ireland) region since the Nextflow AMI was only available for this region. The pipeline was executed using the standard computing environment (no job scheduler) on the EC2. Estimating the cost of running the analysis on the AWS (February 2019 pricing), the m4.10xlarge cost $2.22 per hour7), the standard general purpose solid-state drive (SSD) costs $0.11 per GB-month8. Given that the analysis took approximately 4 hours, the total approximated cost for running the nf-rnaSeqCount pipeline on the SSc data was:
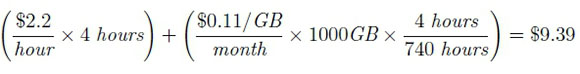
2.5 Benchmarking
The nf-rnaSeqCount pipeline was further benchmarked for time, memory and CPU usage against the popular Rsubread package (Liao et al., 2019). Benchmarking of the nf-rnaSeqCount pipeline against the Rsubread package was to determine the speed at which both tools complete different tasks, computational resource requirements, scalability on large datasets, ability to perform tasks in parallel as well as usability. The Rsubread package was chosen as it is a comprehensive tool and performs similar RNA-seq analysis workflow (read alignment and read counting) as our nf-rnaSeqCount pipeline. The RNA-seq data used for benchmarking was downloaded from the Gene Expression Omnibus (GEO) with an accession GSE1110739. The data consisted of 21 RNA-seq breast cancer samples from walnut-consuming patients (10 samples) and control group (11 samples) (Hardman et al., 2019a, 2019b). Benchmarking of the two tools was carried out on the Wits Computing cluster, with 48 GB of memory and 12 CPUs allocated to each task in the analysis workflow, i.e., reference genome indexing, read alignment and read counting.
Figure 3 summarises the benchmarking results between nf-rnaSeqCount and the Rsubread package in terms of their time, memory and CPU usage when performing genome indexing, read alignment and read counting. The nf-rnaSeqCount pipeline was able to distribute tasks across multiple nodes on the Wits Computing cluster, i.e., run jobs in parallel, in addition to multi-threading, whereas the Rsubread applications were multi-threaded only on a single node.
When it comes to reference genome indexing, both the nf-rnaSeqCount (using STAR and Bowtie) and Rsubread (using index) performed equally in terms of time usage, with each tool completing the task in 66 and 71 minutes, respectively. The nf-rnaSeqCount utilised more resources for indexing (35 GB of memory and 825% of allocated CPUs) compared to Rsubread (16 GB and 99% of allocated CPUs ). For the alignment of the RNA-seq reads to the reference genome, nf-rnaSeqCount (using STAR) outperformed Rsubread (using align), with the alignment tasks completed 31 and 215 minutes, respectively. nf-rnaSeqCount completed the alignment tasks using 30 GB of memory and 648% of the allocated CPUs, whilst Rsubread completed the alignment tasks using 18 GB of memory and 1088% of the allocated CPUs.
Finally, for the read counting, the nf-rnaSeqCount (using htseq-count and featureCounts) was outperformed by Rsubread (using featureCounts), with the read counting tasks completed in 235 and 6 minutes, respectively. nf-rnaSeqCount completed the read counting tasks using 3 GB of memory and 810% of the allocated CPUs, whilst Rsubread completed the alignment tasks using 3 GB of memory and 870% of the allocated CPUs. The huge difference with time usage seen between nf-rnaSeqCount and the Rsubread when it comes to read counting can mainly attributed to htseq-count. Unlike other tools in both workflows, htseq-count cannot be multi-threaded, thus drastically reducing performance and increasing amount of time it takes to complete the read counting tasks for the nf-rnaSeqCount workflow.
3 DISCUSSION
The nf-rnaSeqCount pipeline has been successfully implemented in Nextflow and Singularity, and can be executed on any UNIX-based OS with Nextflow and Singularity installed. It is available on GitHub10 and all the workflow containers with softwares required for running the pipeline are hosted on DockerHub11. In addition to running the pipeline locally, the nf-rnaSeqCount pipeline also supports the PBS and SLURMjob schedulers on HPCs, and this information can be passed to the - profile option of Nextflow when executing the pipeline. Available options are slurm (for SLURM scheduler) and pbs (for PBS scheduler).
The benchmarking results reveal that the nf-rnaSeqCount pipeline compares almost as equally well as the popular Rsubread package in terms of runtime and resource usage. However, nf-rnaSeqCount has added advantages over Rsubread: (1) Parallelisation: in addition to applications being multi-threaded (with the exception of htseq-count) within the workflow, the implementation of nf-rnaSeqCount on Nextflow also allows processes to be run across multiple nodes on HPC, which drastically improves performance when working with large datasets; (2) Installation: there is no need for installation of packages and dependencies for the nf-rnaSeqCount pipeline, e.g., only Nextflow and Singularity are required; and (3) Usability: there is no need for writing tedious scripts for performing RNA-seq analysis with nf-rnaSeqCount, i.e., all inputs, outputs and parameters can be put into a config file which will be used to execute each step of the analysis.
The main requirements for a highly efficient pipeline include reproducibility (capability of the pipeline to reproduce the results under different computational resources), portability (capability of using the pipeline on different computational platforms) and scalability (being able to execute the pipeline on desktop machines, cloud or HPC environments). The nf-rnaSeqCount pipeline presented in this paper meets these requirements for an efficient pipeline. nf-rnaSeqCount is designed on Nextflow and all its application dependencies are packaged in Singularity containers. This makes it possible to run the pipeline on any machine, from desktop to HPC, with both Nextflow and Singularity installed. Nextflow supports a wide variety of job schedulers, and the nf-rnaSeqCount pipeline comes packaged with support for PBS and SLURM schedulers. Advanced users can add their own scheduler support using the nextflow.config file.
The pipeline also comes with detailed documentation on GitHub, for users interested in using this pipeline. The workflow containers hosted on DockerHub ensure that users do not have to go an extra step to install all the softwares required to execute the nf-rnaSeqCount workflow. The modularity of the pipeline not only allows users to interact with results from each step, but also to modify the parameters for the different applications used in the workflow.
4 CONCLUSION
The nf-rnaSeqCount pipeline presented here provides a quick and efficient way to obtain a matrix of read counts (matrix N of n χ m, where is the number of reads assigned to gene i in sequencing experiment j (Rapaport et al., 2013)) that can be used for differential expression and pathway analysis. The output from the nf-rnaSeqCount pipeline can be directly used with popular downstream differential expression analysis tools, such as DESeq2 (Love et al., 2014) and edgeR (Robinson et al., 2010), which take raw read counts as input. The nf-rnaSeqCount pipeline incorporates common tasks associated with RNA-seq data analyses, including QC, read trimming, read alignment and gene quantification. This pipeline largely eliminates the need for multiple scripts and tedious repetitive tasks associated with RNA-seq data analysis, especially when working with large RNA-seq datasets. Users wishing to use the nf-rnaSeqCount pipeline can do so by cloning the repository onto their computational platform (desktop, HPC or cloud) with UNIX-based OS. The availability of workflow containers on DockerHub for executing the nf-rnaSeqCount pipeline eliminates the need for manual installation of applications.
DECLARATIONS
Competing Interests
The authors declare that they have no competing interests.
Funding
This work is based on the research supported in part by the National Research Foundation of South Africa for the Thuthuka Funding Instrument grant (unique grant no. 99206) and Connective Tissue Diseases Research Fund, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg. SH is partially supported by the H3ABioNet grant from the NIH Common Fund Award / NHGRI Grant Number U24HG006941.
Authors' Contributions
P.T. Mpangase: conceptualisation, methodology, software, validation, formal analysis, investigation, data curation, writing (original draft), visualisation, project administration, funding acquisition.
J. Frost: resources, writing (review & editing).
M. Tikly: resources, writing (review & editing).
M. Ramsay: conceptualisation, methodology, writing (review & editing), supervision, funding acquisition.
S. Hazelhurst: conceptualisation, methodology, validation, writing (review & editing), supervision, funding acquisition.
ACKNOWLEDGEMENTS
The authors would like to thank J. Ponomarenko, S. Bonnin, L. Cozzuto, A. Vlasova and P. Di Tommaso from the Centre for Genomic Regulation (CRG) for hosting and assisting P.T. Mpangase; and T. Ngcungcu for kindly allowing us to use the RNA-seq data on albinism for testing the pipeline.
References
Anders, S., Pyl, P. T., & Huber, W. (2015). HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics, 31(2), 166-169. https://doi.org/10.1093/bioinformatics/btu638 [ Links ]
Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. [(01 September 2018, last accessed)]. Retrieved September 1, 2018, from http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Boettiger, C. (2015). An introduction to Docker for reproducible research. ACM SIGOPS Operating Systems Review, 49(1), 71-79. https://doi.org/10.1145/2723872.2723882 [ Links ]
Bolger, A. M., Lohse, M., & Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30(15),2114-2120. https://doi.org/10.1093/bioinformatics/btu170 [ Links ]
Conesa, A., Madrigal, P., Tarazona, S., Gomez-Cabrero, D., Cervera, A., McPherson, A., Szczes-niak, M. W., Gaffney, D. J., Elo, L. L., Zhang, X., & Mortazavi, A. (2016). A survey of best practices for RNA-seq data analysis. Genome Biology, 17(1), 13. https://doi.org/10.1186/s13059-016-0881-8 [ Links ]
Di Tommaso, P., Chatzou, M., Floden, E. W., Barja, P. P., Palumbo, E., & Notredame, C. (2017). Nextflow enables reproducible computational workflows. Nature Biotechnology, 35(4), 316-319. https://doi.org/10.1038/nbt.3820 [ Links ]
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M., & Gingeras, T. R. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29(1), 15-21. https://doi.org/10.1093/bioinformatics/bts635 [ Links ]
Ewels, P., Magnusson, M., Lundin, S., & Kaller, M. (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 32(19), 3047-3048. https://doi.org/10.1093/bioinformatics/btw354 [ Links ]
Fan, J., Han, F., & Liu, H. (2014). Challenges of Big Data analysis. National Science Review, 1(2), 293-314. https://doi.org/10.1093/nsr/nwt032 [ Links ]
Finotello, F., & Di Camillo, B. (2015). Measuring differential gene expression with RNA-seq: challenges and strategies for data analysis. Briefings in Functional Genomics, 14(2), 130142. https://doi.org/10.1093/bfgp/elu035 [ Links ]
Frost, J., Estivill, X., Ramsay, M., & Tikly, M. (2019). Dysregulation of the Wnt signaling pathway in South African patients with diffuse systemic sclerosis. Clinical Rheumatology, 38(3), 933-938. https://doi.org/10.1007/s10067-018-4298-5 [ Links ]
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., Adiconis, X., Fan, L., Raychowdhury, R., Zeng, Q., Chen, Z., Mauceli, E., Hacohen, N., Gnirke, A., Rhind, N., di Palma, F., Birren, B. W., Nusbaum, C., Lindblad-Toh, K., ... Regev, A. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology, 29(7), 644-652. https://doi.org/10.1038/nbt.1883 [ Links ]
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., Couger, M. B., Eccles, D., Li, B., Lieber, M., MacManes, M. D., Ott, M., Orvis, J., Pochet, N., Strozzi, F., Weeks, N., Westerman, R., William, T., Dewey, C. N., . Regev, A. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols, 8(8), 1494-1512. https://doi.org/10.1038/nprot.2013.084 [ Links ]
Hardman, W. E., Primerano, D. A., Legenza, M. T., Morgan, J., Fan, J., & Denvir, J. (2019a). Dietary walnut altered gene expressions related to tumor growth, survival, and metastasis in breast cancer patients: a pilot clinical trial. Nutrition Research, 66, 82-94. https://doi.org/10.1016/j.nutres.2019.03.004 [ Links ]
Hardman, W. E., Primerano, D. A., Legenza, M. T., Morgan, J., Fan, J., & Denvir, J. (2019b). mRNA expression data in breast cancers before and after consumption of walnut by women. Data in Brief, 25, 104050. https://doi.org/10.1016/j.dib.2019.104050 [ Links ]
Kluge, M., & Friedel, C. C. (2018). Watchdog - a workflow management system for the distributed analysis of large-scale experimental data. BMC Bioinformatics, 19(1), 97. https://doi.org/10.1186/s12859-018-2107-4 [ Links ]
Kurtzer, G. M., Sochat, V., & Bauer, M. W. (2017). Singularity: Scientific containers for mobility of compute (A. Gursoy, Ed.). PLOS ONE, 12(5), e0177459. https://doi.org/10.1371/journal.pone.0177459 [ Links ]
Langmead, B., Trapnell, C., Pop, M., & Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology, 10(3), R25. https://doi.org/10.1186/gb-2009-10-3-r25 [ Links ]
Li, B., Fillmore, N., Bai, Y., Collins, M., Thomson, J. A., Stewart, R., & Dewey, C. N. (2014). Evaluation of de novo transcriptome assemblies from RNA-Seq data. Genome Biology, 15(12), 553. https://doi.org/10.1186/s13059-014-0553-5 [ Links ]
Liao, Y., Smyth, G. K., & Shi, W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30(7), 923-930. https://doi.org/10.1093/bioinformatics/btt656 [ Links ]
Liao, Y., Smyth, G. K., & Shi, W. (2019). The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Research, 47(8), e47-e47. https://doi.org/10.1093/nar/gkz114 [ Links ]
Love, M. I., Huber, W., & Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology, 15(12), 550. https://doi.org/10.1186/s13059-014-0550-8 [ Links ]
Perkel, J. (2016). Democratic databases: science on GitHub. Nature, 538(7623), 127-128. https://doi.org/10.1038/538127a [ Links ]
Piccolo, S. R., & Frampton, M. B. (2016). Tools and techniques for computational reproducibility. GigaScience, 5(1), 30. https://doi.org/10.1186/s13742-016-0135-4 [ Links ]
Rapaport, F., Khanin, R., Liang, Y., Pirun, M., Krek, A., Zumbo, P., Mason, C. E., Socci, N. D., & Betel, D. (2013). Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biology, 14(9), R95. https://doi.org/10.1186/gb-2013-14-9-r95 [ Links ]
Robinson, M. D., McCarthy, D. J., & Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26(1), 139-140. https://doi.org/10.1093/bioinformatics/btp616 [ Links ]
Schulz, W., Durant, T., Siddon, A., & Torres, R. (2016). Use of application containers and workflows for genomic data analysis. Journal of Pathology Informatics, 7(1), 53. https://doi.org/10.4103/2153-3539.197197 [ Links ]
Trapnell, C., Hendrickson, D. G., Sauvageau, M., Goff, L., Rinn, J. L., & Pachter, L. (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nature Biotechnology, 31(1), 46-53. https://doi.org/10.1038/nbt.2450 [ Links ]
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., Pimentel, H., Salzberg, S. L., Rinn, J. L., & Pachter, L. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols, 7(3), 562-578.https://doi.org/10.1038/nprot.2012.016 [ Links ]
Received: 26 May 2020
Accepted: 17 August 2021
Available online: 19 December 2021
Email: Phelelani T. Mpangase Phelelani.Mpangase@wits.ac.za (corresponding), Jacqueline Frost jacqueline.frost@mssm.edu, Mohammed Tikly Mohammed.Tikly@wits.ac.za, Michèle Ramsay Michele.Ramsay@wits.ac.za, Scott Hazelhurst Scott.Hazelhurst@wits.ac.za
1 http://docker.io/
2 https://www.sylabs.io/
3 http://github.com/
4 https://hub.docker.com/
5 http://hpc.uct.ac.za/
6 https://aws.amazon.com/
7 https://aws.amazon.com/ec2/pricing/on-demand/
8 https://aws.amazon.com/ebs/pricing/
9 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE111073
10 https://github.com/phelelani/nf-rnaSeqCount
11 https://hub.docker.com/r/phelelani/nf-rnaseqcount


{kind=link}
{kind=link}
{kind=link}