










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkJournal of the Southern African Institute of Mining and Metallurgy
On-line version ISSN 2411-9717
Print version ISSN 2225-6253
J. S. Afr. Inst. Min. Metall. vol.118 n.12 Johannesburg Dec. 2018
http://dx.doi.org/10.17159/2411-9717/2018/v118n12a4
PAPERS OF GENERAL INTEREST
Decomposition of hydrogen peroxide in alkaline cyanide solutions
T. FungeneI; D.R. GrootI, II; T. MahlanguIII; K.C. SoleI
IDepartment of Materials Science and Metallurgical Engineering, University of Pretoria, South Africa
IIDepartment of Mining and Process Engineering, Namibia University of Science and Technology, Windhoek, Namibia
IIIRandgold Resources, Johannesburg, South Africa
SYNOPSIS
Although oxygen is widely employed as the oxidant of choice in gold leaching by cyanide, its low aqueous solubility presents some drawbacks in practical application; hydrogen peroxide has therefore been considered as a possible alternative. The aim of this investigation was to study the catalytic decomposition of hydrogen peroxide, which generates an oxidizing intermediate species, and to understand its effect on cyanide destruction. Operating conditions that facilitated the effective decomposition of hydrogen peroxide were established by varying the pH and catalyst type and concentration. The oxidizing intermediate, detected using an indirect technique, was found to be the hydroxyl radical (OH). OH' is commonly generated in acidic solutions, but this work demonstrated that it is also produced at the alkaline pH values necessary for cyanide gold leaching. The effects of free and complexed iron and copper catalysts on the oxidation and consumption of hydrogen peroxide and cyanide were also investigated. It was shown that the cyano complexes of Fe(II) and Cu(I) are also effective as decomposition catalysts. Hydrogen peroxide concentrations above 0.01 M decreased the free cyanide concentration, which was attributed to the probable formation of the cyanate anion (CNO-). Although cyanide consumption increased due to its oxidation in the presence of OH', excessive cyanide consumption in the presence of copper was attributed primarily to its complexation by the unstable copper(I) cyanide species. Rate constants for the decompositions of H2O2 and cyanide by ferrocyanide and copper cyanide were calculated; the latter was identified as being a better catalyst.
Keywords: hydrogen peroxide, decomposition, Fenton chemistry, cyanide, gold leaching, radical.
Introduction
Leaching of gold from its ores using cyanide has been employed for well over a century (Habashi, 2016). Addition of cyanide converts the gold into a cyanide complex (Au(CN)2) that is soluble in water (Wang and Forssberg, 1990). The leaching reaction can be represented as Equation [1]:
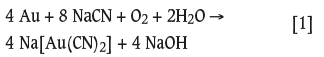
As shown above, leaching also requires the presence of an oxidant, so a source of oxygen (typically air) is introduced into the leach pulp. Leach kinetics are, however, limited by the slow rate at which oxygen transfers from the gaseous to the liquid phase and the resulting low levels of dissolved oxygen. Even with the use of enhanced aeration techniques, such as compressed air or pure oxygen (Adams, 2016; Loroesch, 1990), the maximum aqueous solubility of dissolved oxygen is about 20 mg/L (Loroesch, 1990). To overcome this limitation of oxygen mass transfer, the application of a liquid oxidant, such as hydrogen peroxide (H2O2), has been proposed (Ball et al., 1989; Knorre et al., 1993, 1994).
Use of H2O2 in the cyanide leaching of gold has the potential to ensure the fast and homogenous distribution of an active oxygen species in the pulp. Peroxide-assisted leaching has been acknowledged to exhibit enhanced kinetics and improved gold recoveries compared with both conventional and improved aeration techniques (Arslan et al., 2003; Guzman et al., 1999; Loroesch, 1990); however, these advantages need to be weighed against the increased cyanide consumption due to its loss by oxidation by H2O2.
When H2O2 is added to an aqueous system, the resulting decomposition produces dissolved oxygen that is directly available for leaching:
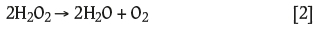
This decomposition is well known to be catalysed by metallic species, specifically iron and copper, in what are termed Fenton and Fenton-like reactions, respectively (Fenton, 1894; Haber and Weiss, 1934; Watts and Teel, 2005). H.J.H. Fenton discovered in 1894 that several metals exhibit a strong catalytic effect that generates highly reactive hydroxyl radicals; these impart oxygen transfer properties that improve the use of hydrogen peroxide. Iron and copper commonly occur as impurities in gold ores (usually as sulphides), so leaching can benefit from the enhanced dissolved oxygen content resulting from the catalytic dissociation of H2O2into free oxygen and water. However, if the pH is too high, iron will precipitate as Fe(OH)3 and H2O2 will decompose to oxygen.
Debate regarding the reaction pathway for the decomposition of H2O2, specifically concerning the nature of the oxidizing intermediate(s), is ongoing (Barbusinski, 2009). Two main pathways are postulated: one considers hydroxyl radical (OH') formation (Barbusinski, 2009; Deguillaume et al., 2005; Haber and Weiss, 1932, 1934); the other is a non-radical pathway that considers ferryl ion (FeO2+) formation (Barbusinski, 2009; Bray and Gorin, 1932). The respective reactions are shown in Equations [3] and [4]:
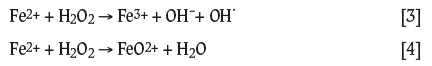
An aim of this work was to determine the pathway for the decomposition of H2O2, i.e., to identify whether a radical is involved in the reaction, specifically in alkaline cyanide solutions. This could lead to a better understanding of the mechanism by which dissolved oxygen levels are improved by the addition of H2O2 and its effects on other possible consumers of cyanide (CN-) in gold leaching.
The study comprised four main parts: determination of the effects of pH and catalyst type and concentration on the decomposition of H2O2; the effect of H2O2 concentration on the decomposition of cyanide; examination of Fenton and Fenton-like reactions in the presence of cyanide; and detection of the presence of radicals in acidic and alkaline solutions.
Experimental
Reagents
Hydrogen peroxide (30%) was employed as received. Copper and iron, employed as the decomposition catalysts, were added as the respective sulphate salts for experiments in the absence of cyanide and as K4Fe(CN)6 and CuCN for those in the presence of cyanide. Cyanide was also provided as NaCN in some experiments. Concentrated (98%) H2SO4 and CaO were employed for pH adjustment. All chemicals were of analytical reagent grade, supplied by Merck and Sigma Aldrich.
Methods
Effect ofpH and catalyst concentration on the decomposition of H2O2
The experiments for each catalyst were carried out at both acidic (the natural pH of the catalyst, defined as the resulting pH when the metal salt was added to water: pH 2-3 for iron; pH 3-4 for copper) and alkaline conditions (pH 9-10 obtained by addition of CaO). Sixty millilitres (mL) of H2O2 (0.6 M) and 0.5 g/L (approx. 0.01 M) of iron or copper, obtained by addition of the respective sulphate salt, were added to deionized water to make up a volume of 1000 mL. The solutions were magnetically stirred at a constant speed of 500 r/min. Individual experiments were carried out at pH values ranging from 4 to 11. After 15 minutes at each pH, a 10 mL aliquot was analysed for residual H2O2 concentration.
To study the effect of catalyst concentration, experiments were carried out using an initial metal catalyst concentration of 0.1 g/L (approx. 0.002 M). This was increased stepwise to 3 g/L (approx. 0.06 M) after equilibration for 15 minutes. After equilibration, the dissolved oxygen (DO) content, redox potential (Eh) (reported relative to the Ag/AgCl reference electrode, E0= 0.222 V), and H2O2 content of each solution were measured.
Effect of H2O2concentration on the decomposition of cyanide
The effect of H2O2 concentration on cyanide decomposition in the absence of a metal catalyst was determined using 1000 mL of 0.015 M NaCN and initial H2O2 concentrations ranging from 0.005 M to 0.1 M. CaO was added to maintain a pH of 10-11.5 throughout each test. Samples taken after selected reaction times were analysed for pH and cyanide content.
Effect of iron and copper (free and complexed) on the decomposition of H2O2and cyanide
The effects of iron and copper (as both free and complexed ions) on H2O2 decomposition were investigated to determine whether H2O2 would decompose similarly in both Fenton and Fenton-like reactions. The metals were added both as sulphates (FeSO4-7H2O; CuSO4-5H2O) and as cyanide complexes (K4Fe(CN)6; CuCN) at a concentration of 0.004 M to a solution of 0.01 M H2O2. The pH was maintained in the range 10-11.5.
Determination of the effects of iron and copper cyanide complexes on the aqueous free cyanide concentration followed the same experimental procedure as described above, with 0.01 M free cyanide added as NaCN. The tests were carried out with and without the addition of 0.01 M H2O2. Samples taken at selected reaction times were analysed for pH, H2O2, and cyanide content.
Radical detection studies
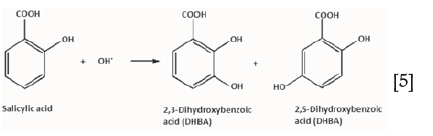
An indirect method for identifying the presence of the hydroxyl radical was employed, based on analysis of the products of the reaction between salicylic acid and OH'. Detection of the 2,3- or 2,5-dihydroxybenzoic acid (DHBA) isomers (Equation [5]) confirms the presence of OH' (Nguyen et al., 2008). Fenton reactions were carried out in the presence of sufficient salicylic acid to react with the free OH' generated. To a 1 L solution of specified pH (acidic or alkaline), 0.01 M Fe2+ was added, followed by 0.5 M H2O2 and 0.12 M salicylic acid (stoichiometric excess of 20% with respect to Fe2+). The solution was allowed to react for 1 hour at 30°C and then cooled to room temperature, after which it was poured into a separating funnel and intimately mixed with diethyl ether to extract only the desired organic products (2,3- and/or 2,5-DHBA). The organic phase was evaporated over low heat, leaving a solid that was collected for identification by infrared (IR) and nuclear magnetic resonance (NMR) spectroscopy.
Analytical methods
H2O2 concentrations were titrimetrically determined using 0.025 M KMnO4 (Klassen et al, 1994). Free cyanide (CN-) was determined by titration with 0.1 M AgNO3, using Rhodamine B as the indicator (Mendham et al., 2000). DO content was measured using a dissolved oxygen meter (model 5100 OUR/SOUR, YSI, USA); pH and Eh were measured using a combination meter (model 704, Metrohm, Switzerland). All tests were repeated twice, giving a total of three replicates to establish the measure of uncertainty, indicated by the error bars on the graphs in Figures 1 to 9.



Results and discussion
Effect ofpH on decomposition ofhydrogen peroxide
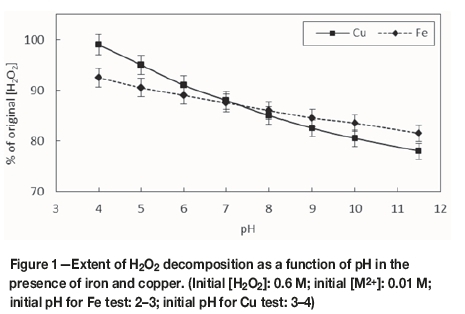
Figure 1 shows the effect of equilibrium pH on the extent of decomposition of H2O2 in the presence of 0.01 M iron or copper. It is evident that, for both metals, an increase in pH increased the extent of decomposition. This result is in agreement with those of Nicoll and Smith (1955), who observed that the decomposition of H2O2 in distilled water in the presence of a catalytic impurity increased as the alkalinity of the solution increased. The effective decomposition at high pH values accounts for the reported efficiency of H2O2 as an oxidant in alkaline gold leaching (Guzman et al., 1999). It is also evident that copper resulted in more effective decomposition of H2O2 above pH 7, indicating that it is a better catalyst than iron in the alkaline pH range.
Effect ofcatalyst concentration on decomposition of H2O2 in acidic and alkaline solutions
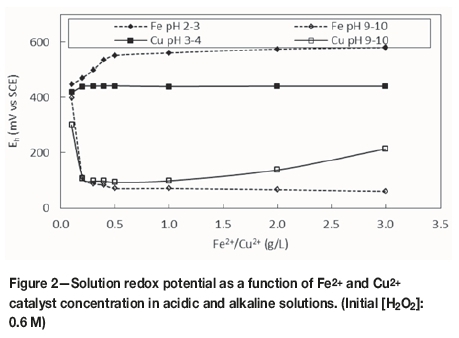
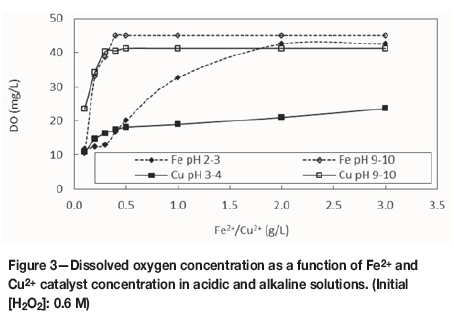
Figures 2 and 3 compare the effects of increasing iron or copper catalyst concentration on the Eh and DO due to H2O2 decomposition in acidic (natural pH for each catalyst) and alkaline (pH 9-10) ranges.
Figure 2 shows that the solution potential gradually increased at acidic pH values for increased iron concentrations because the ferrous iron concentration decreased as a result of oxidation to ferric ions in the presence of H2O2:
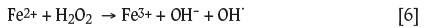
This effect was not evident for Cu2+, which is stable in the divalent form under these conditions. In contrast, a decrease in Eh was observed in alkaline solutions for both metals. This is attributed to the formation of stable hydroxide species of both iron and copper under these conditions. These species are known to be active catalysts in the decomposition of H2O2 (Lin and Gurol, 1998). At alkaline pH, heavy metal ions (such as iron or copper) form unstable peroxides, causing decomposition of H2O2; in addition, colloidal hydroxides are formed as the pH increases. These metal hydroxides are considered to be more active catalysts for H2O2 decomposition than the free or complexed metal ions (Lin and Gurol, 1998; Nicoll and Smith, 1955). It is notable that both catalysts had the most significant effect at a concentration of 0.5 g/L; above this concentration, further improvements in performance were limited.
As shown in Figure 3, the DO content was higher in alkaline solutions for catalyst concentrations above 0.5 g/L, which indicates greater extents of decomposition of H2O2 (see Equation [2]). This confirmed that such stable hydroxides tend to be better catalysts than their respective free transition-metal cations (Lin and Gurol, 1998; Nicoll and Smith, 1955). It is also notable that the DO concentrations reached close to 50 mg/L under these conditions, which is considerably higher than the equilibrium oxygen concentration reported when using enhanced aeration with gaseous oxygen (approx. 20 mg/L) (Loroesch, 1990).
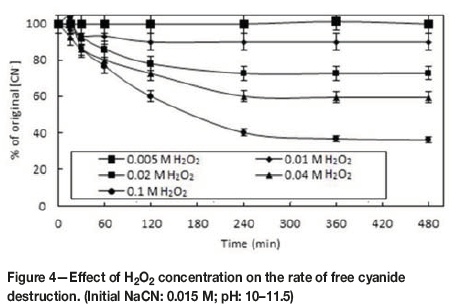
Effect ofH2O2 concentration on rate of decomposition ofcyanide
The use of H2O2 in treating wastewaters containing cyanide and/or cyanide complexes is common practice (Ozcan et al., 2011). This ability of H2O2to destroy cyanide potentially mitigates against its use in cyanide leaching systems. Figure 4 shows the effect of H2O2 concentrations ranging from 0.005 M to 0.10 M on the rate of decomposition of free cyanide. A significant decrease in CN- concentration was observed for H2O2 concentrations above 0.01 M. These results agree with those of similar work carried out by Guzman et al. (1999), who attributed the decrease in cyanide concentration to the formation of the cyanate anion (CNO-), according to Equation [7]:
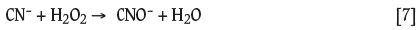
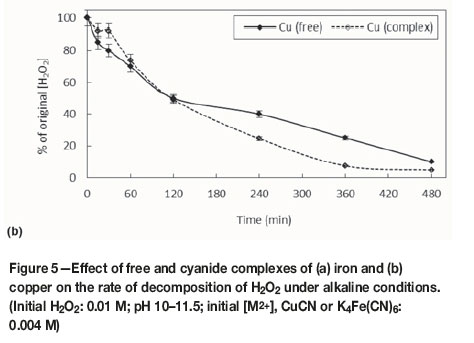
Effect ofiron and copper (free and complexed) on decomposition rate of H2O2
Figures 5a and 5b show the effects of iron and copper, respectively, added as sulphate and complexed cyanide salts, on the rate of H2O2 decomposition. Both the free and complexed cations affected the stability of H2O2. The rate of decomposition in the presence of Fe was essentially unaffected by the nature of the ion; copper, however, had a more pronounced effect when present as the cyanide complex. Although copper(II)-cyano complexes have been characterized, they are unstable and decompose rapidly, forming copper(I)-cyano complexes and cyanate, CNO-, in the presence of H2O2 (Sceresini and Breuer, 2016): the presence of cupric copper (the free Cu2+ ion) therefore causes loss of cyanide as cyanate. These results indicated that H2O2 decomposed in the presence of iron and copper as both free and complexed species in Fenton and Fenton-like reactions, respectively.
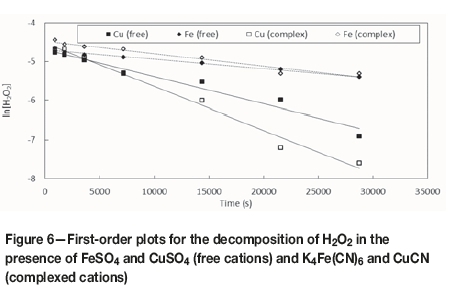
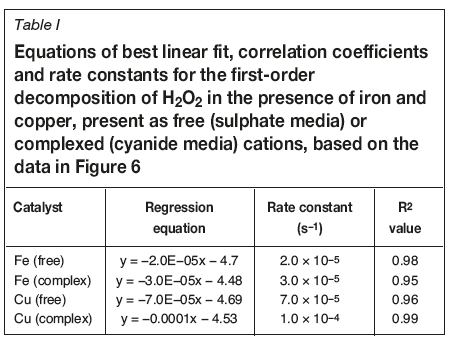
Figure 6 shows the corresponding first-order plots based on the data of Figure 5. The rate constants, k, for the decomposition of H2O2 by these catalysts were calculated from the slope of a linear least-squares fit:
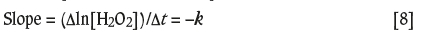
where the concentration of H2O2 is given in M.
The equations, rate constants and correlation coefficients (R2) for the linear plots are summarized in Table I. The relative values of the rate constants confirm that the rate of decomposition of H2O2 in the presence of copper was faster than that of iron and that the rates were slightly faster when the metal ions were present in the form of the respective cyanide complexes. The free copper ion reduced the initial H2O2 concentration by 90% in 480 minutes, compared with only 55% in the presence of the free iron species. Similarly, the presence of copper cyanide reduced the H2O2 concentration by 95% in 480 minutes, compared with only 50% for ferrocyanide (Figure 6).
Effect of iron and copper cyanide complexes on rate ofdecomposition offree cyanide in the absence and presence H2O2
Using a H2O2 concentration of 0.01 M, which was shown to have negligible effect on the degradation of free cyanide (supplied as NaCN) (Figure 4), it was of interest to monitor whether cyanide would be similarly destroyed when the catalysts were introduced as the cyano complex ions. Figures 7a and 7b show the decrease in free cyanide with time in the presence and absence of H2O2 when using iron and copper as the catalysts, respectively. Ferrocyanide is regarded as a strongly bound complex and is therefore not easily dissociated (Adams, 2016; Sharpe, 1976), which is why it tends to remain stable during detoxification of free cyanide and cyanide complexes by H2O2 (Griffiths et al., 1987; Ozcan et al., 2011). A small decrease in the initial free cyanide concentration was, however, still observed (Figure 7a) This was attributed to a Fenton-like reaction between H2O2 and ferrocyanide, which produces the hydroxyl radical intermediate (OH') that, in turn, reacts with free CN-. In the absence of H2O2, no decrease in the free cyanide concentration occurred for the case of ferrocyanide, confirming the stability of the complex.
For the case of CuCN, the free cyanide concentration dropped markedly both in the presence and absence of H2O2. It is well-known, however, that copper forms a series of strong cyanide complexes (Ringbom, 1963), of which the tri-and tetracyanide species predominate at alkaline pH values (Sceresini and Breuer, 2016):
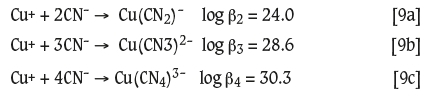
Only a slightly greater extent of cyanide destruction occurred in the presence of H2O2, indicating that this phenomenon is primarily due to complexation, rather than to reaction with the hydroxyl radical due to Fenton-like reactions.
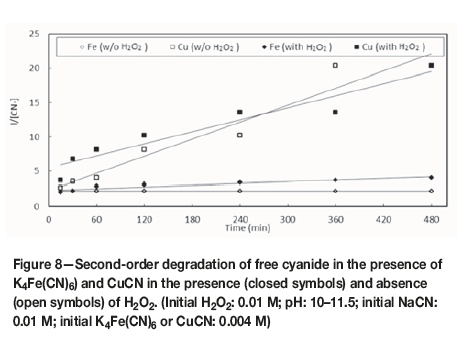
The degradation of free cyanide both with and without H2O2 occurred according to second-order kinetics in the presence of iron and copper cyanides, as shown in Figure 8, derived from the data presented in Figure 7. The relative values of the rate constants (Table II), calculated by the equation:
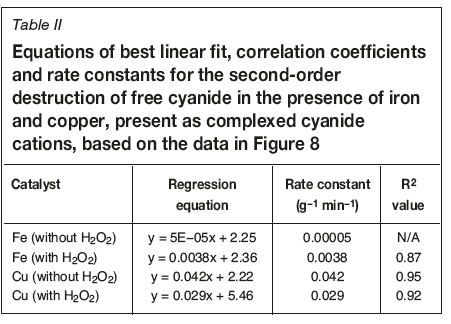

where the concentration of CN- was calculated in g/L, confirmed that copper(I) cyanide caused faster decomposition of CN- than ferrocyanide, both with and without H2O2.
Radical detection studies
Salem et al., (2000) proposed OH' as an intermediate product in reactions where the catalyst is a transition-metal complex. In this work, evidence for the existence of the OH' radical in the decomposition of H2O2 was assumed based on an indirect method (Nguyen et al., 2008). Aromatic hydroxylation of salicylic acid to specifically yield the 2,3- and/or 2,5-DBHA isomers can only occur via the OH' radical (see Equation [5]). The hydroxylation reaction was carried out in the presence of H2O2 and an iron catalyst under acidic pH (2-3) and alkaline pH (10-11) conditions and the reaction products were analyzed.
NMR spectra of the hydroxylation products compared well with that of the standard 2,3-DHBA isomer (National Institute of Advanced Industrial Science and Technology, 2013): peaks characteristic of the hydrogen atoms of this isomer were observed at chemical shifts of 6.5 to 8.5 ppm. IR spectra indicated the presence of carboxylic acid, alcohol, and carbonyl groups, which are the functional groups present in this isomer. Identification of the hydroxylation product of salicylic acid as 2,3-DHBA by NMR and IR spectroscopy, irrespective of whether the reaction was carried out in acidic or alkaline media, indirectly confirmed the presence of OH' and showed that H2O2 decomposed to this radical under both pH conditions. It was important to confirm the presence of this intermediate because it is highly reactive (standard reduction potential of 1.4 V) compared with the ferryl ion (0.9 V) (Petri et al., 2011) and could possibly react with contaminants in a gold leach pulp, leading to further losses in such application. This high reduction potential of OH' may also improve Au leaching kinetics.
Conclusions
The aim of this work was to study how the addition of H2O2 affects the cyanide concentration, the Eh, and the DO concentration in cyanide gold leaching. The effects of transition-metal cation catalysts, such as iron and copper (free and complexed), and pH on the stability of H2O2 were established. It was found that the cyano complexes of Fe(II) and Cu(I) effectively act as hydrogen peroxide decomposition catalysts in alkaline media. Increasing pH and catalyst concentration increased the rate of decomposition of H2O2. Copper (free and complexed) cyanide was found to be more effective in decomposing H2O2 than the corresponding iron(II) species.
It was established that a H2O2 concentration greater than 0.01 M caused loss of free cyanide. In the presence of copper, loss of free cyanide by complexation was attributed to the formation of stable higher copper(I) cyanide complexes. No loss of cyanide by complexation was observed in the presence of ferrocyanide, in accordance with the known relatively high stability of this complex. Additional losses of cyanide in the presence of hydrogen peroxide were attributed to the presence of the hydroxyl radical in Fenton-like reactions for both iron and copper.
An oxidizing intermediate in the Fenton and Fenton-like reactions, assumed on the basis of detection and identification by an indirect technique to be the hydroxyl radical, was found to be present in both acidic and alkaline solutions. This radical was responsible for the increased oxidation potential observed in alkaline solutions. Proving the presence of this species in alkaline solutions (in the presence of transition metal ion complexes) and understanding its effect on free cyanide degradation could aid in minimizing cyanide losses and establishing economical H2O2 dosages for industrial applications in gold leaching.
Acknowledgements
The authors are grateful for financial support from the South African Minerals to Metals Research Institute.
References
Adams, M.D. (ed.) 2016. Gold Ore Processing. Project Development and Operation. 2nd edn. Elsevier, Oxford. [ Links ]
Arslan, F., Ozdamar, D.Y., and Muduroglu, M. 2003. Cyanidation of Turkish gold-silver ore and the use of hydrogen peroxide. European Journal of Mineral Processing and Environmental Protection, vol. 3, no. 3. pp. 309-315. [ Links ]
Ball, S.P., Monhemius, A.J., and Wyborn, P.J. 1989. The use of inorganic peroxides as accelerators for gold heap leaching. Proceedings of Precious Metals 89. Jha, M.C. and Hill, S.D. (eds.). The Minerals, Metals and Materials Society, Warrendale, PA. pp. 149-164. [ Links ]
Barbusinski, K. 2009. Fenton reaction-controversy concerning the chemistry. Ecological Chemistry and Engineering, vol. 16, no. 3. pp. 347-358. [ Links ]
Bray, W.C. and Gorin, M.H. 1932. Ferryl ion, a compound of tetravalent iron. Journal of the American Chemical Society, vol. 54, no. 5. pp. 2124-2125. [ Links ]
Deguillaume, L., Leriche, M., and Chaumerliac, N. 2005. Impact of radical versus non-radical pathway in the Fenton chemistry on the iron redox cycle in clouds. Chemosphere, vol. 6, no. 5. pp. 718-724. [ Links ]
Fenton, H.J.H. 1894. LXXIII.-Oxidation of tartaric acid in presence of iron. Journal of the Chemical Society, Transactions, vol. 65. pp. 899-910. [ Links ]
Griffiths, Α., Knorre, H., Gos, S., and Higgins, R. 1987. The detoxification of gold-mill tailings with hydrogen peroxide. Journal of the South African Institute of Mining and Metallurgy, vol. 87, no. 9. pp. 279-283. [ Links ]
Guzman, L., Segarra, M., Chimenos, J.M., Fernandez, M.A., and Espiell, F. 1999. Gold cyanidation using hydrogen peroxide. Hydrometallurgy, vol. 52, no. 1. pp. 21-35. [ Links ]
Habashi, F. 2016. Gold - an historical introduction. Gold Ore Processing: Project Development and Operations. 2nd edn. Adams, M.D. (ed.). Elsevier, Oxford. pp. 1-24. [ Links ]
Haber, F. and Weiss, J. 1932. Über die katalyse des hydroperoxydes. Naturwissenschaften, vol. 20, no. 51. pp. 948-950. [ Links ]
Haber, F. and Weiss, J. 1934. The catalytic decomposition of hydrogen peroxide by iron salts. Proceedings of the Royal Society of London A: Mathematical, Physical and Engineering Sciences, vol. 147, no. 861. pp. 332-351. [ Links ]
Klassen, N.V., Marchington, D., and McGowan, H.C. 1994. H2O2 determination by the I3-method and by KMnO4 titration. Analytical Chemistry, vol. 66, no. 18. pp. 2921-2925. [ Links ]
Knorre, H., Loroesch, J., Gos, S., Stoll, M., and Ziegler, Α. 1993. Process for leaching precious metals with hydrogen peroxide and cyanide leaching solution. US patent 5250272. [ Links ]
Knorre, H., Griffiths, Α., Loroesch, J., and Fischer, J. 1994. Process for the leaching of gold and silver cyanide leaching solution and controlled addition of hydrogen peroxide. US patent 5275791. [ Links ]
Lin, S.S. and Gurol, M.D. 1998. Catalytic decomposition of hydrogen peroxide on iron oxide: kinetics, mechanism, and implications. Environmental Science and Technology, vol. 32, no. 10. pp. 1417-1423. [ Links ]
Loroesch, J. 1990. Peroxide-assisted leach: three years of increasing success. Proceedings of the Randol Gold Forum '90. Randol International Ltd., Golden, CO. pp. 215-20. [ Links ]
Mendham, J., Denney, R.C., Barnes, J.D., and Thomas, M. 2000. Titrimetric analysis. Vogels Textbook of Quantitative Chemical Analysis. 6th edn. Pearson Education Ltd., Harlow, Prentice Hall. pp. 447-449. [ Links ]
National Institute of Advanced Industrial Science and Technology (AIST). Spectral Database for Organic Compounds. Japan. http://sdbs.db.aist.go.jp/sdbs/cgi-bin/cre_index.cgi. [accessed April 2013]. [ Links ]
Nguyen, V., Bonds, D.V., and Prokai, L. 2008. Measurement of hydroxyl-radical formation in the rat striatum by in vivo microdialysis and GC-MS. Chromatographia, vol. 68, no. 1. pp. 57-62. [ Links ]
Nicoll, W.D. and Smith, A.F. 1955. Stability of dilute alkaline solutions of hydrogen peroxide. Industrial and Engineering Chemistry, vol. 47, no. 12. pp. 2548-2554. [ Links ]
Ozcan, E., Gok, Z., and Yel, E. 2011. Photo/photochemical oxidation of cyanide and metal-cyanide complexes: ultraviolet Α versus ultraviolet C. Environmental Technology, vol. 33, no. 16-18. pp. 1913-1925. [ Links ]
Petri, B.G., Watts, R.J., Teel, A.L., Huling, S.G., and Brown, R.A. 2011. Fundamentals of ISCO using hydrogen peroxide. In Situ Chemical Oxidation/or Groundwater Remediation. 1st edn. Siegrist, R.L., Crimi, M., and Simpkin, T.J. (eds.). Springer, New York. pp. 33-88. [ Links ]
Ringbom, Α. 1963. Complexation in Analytical Chemistry. Interscience Publishers, New York. [ Links ]
Salem, I.A., El Maazawi, M., and Zaki, A.b. 2000. Kinetics and mechanisms of decomposition reaction of hydrogen peroxide in presence of metal complexes. International Journal of Chemical Kinetics, vol. 32, no. 11. pp. 643-666. [ Links ]
Sceresini, B. and Breuer, P. 2016. Copper-gold ores. Gold Ore Processing: Project Development and Operations. 2nd edn. Adams, M.D. (ed.). Elsevier, Oxford. pp. 771-801. [ Links ]
Sharpe, A.G. 1976. Chemistry of Cyano Complexes of the Transition Metals. Academic Press, London. [ Links ]
Wang, X., and Forssberg, K.E. 1990. The chemistry of cyanide-metal complexes in relation to hydrometallurgical processes of precious metals. Mineral Processing and Extractive Metallurgy Review, vol. 6. pp. 81-125. [ Links ]
Watts, R.J. and Teel, A.L. 2005. Chemistry of modified Fenton's reagent (catalyzed H2O2 propagations-CHP) for in situ soil and groundwater remediation. Journal of Environmental Engineering, vol. 131, no. 4. pp. 612-622. [ Links ] ♦
Paper received Nov. 2017
Revised paper received Mar. 2018

