










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkJournal of the Southern African Institute of Mining and Metallurgy
versión On-line ISSN 2411-9717
versión impresa ISSN 2225-6253
J. S. Afr. Inst. Min. Metall. vol.118 no.4 Johannesburg abr. 2018
http://dx.doi.org/10.17159/2411-9717/2018/v118n4a7
PAPERS OF GENERAL INTEREST
Density functional theory prediction of the aqueous speciation of ZrF4 and HfF4
D.B. Jansen van Vuuren; H.M. Krieg; D.J. van der Westhuizen; C.G.C.E. van Sittert
North-West University, Potchefstroom Campus, Potchefstroom, South Africa
SYNOPSIS
Computational chemistry was used to make predictions regarding the stepwise hydrolysis and fluoride dissociation reactions of ZrF4 and HfF4 in aqueous solutions. Specifically, density functional theory was used to predict Gibbs formation energies for ZrF4 and HfF4 in aqueous solutions, which were compared to experimental results reported in the literature. Following this, Gibbs reaction energies for the stepwise hydrolysis and fluoride dissociation reactions of ZrF4 and HfF4 were predicted, also with density functional theory, and used to predict stability constants that describe the distribution of the various zirconium and hafnium species in an aqueous solution. Finally, given these stability constants, distribution of species diagrams were constructed, which display which ZrF4 and HfF4 species are present at a given pH.
Keywords: Speciation, ZrF4, HfF4, density functional theory, stability constant.
Introduction
Zirconium (Zr) and hafnium (Hf have important applications in the nuclear energy industry, where Zr is used as part of the cladding material of the uranium fuel rod tubes that are used during the nuclear reactions, while Hf is used in the control rods that sometimes moderate these reactions (Banda and Lee, 2015). Since Zr and Hf occur together in nature they have to be separated prior to use in nuclear reactors (Blumenthal, 1958). A promising technology that can be used to separate these metals is hydrometallurgical solvent extraction (SX) (Banda and Lee, 2015). In order to understand the mechanisms that underpin the SX of Zr and Hf it is vital to know which Zr and Hf complexes are dominant in the aqueous phase.
It is well established that solutions of Zr and Hf chlorides and oxychlorides are dominated by tetrameric complexes (Hu et al., 2013; Kalaji et al., 2014; Hagfeldt, Kessler, and Persson, 2004). However, as the concentration of certain strongly coordinating ligands such as sulphate or nitrate anions increases, the polymerization of Zr and Hf complexes in chloride and oxychloride solutions tends to decrease. For instance, recent studies of Zr and Hf chloride and perchlorate solutions using high-energy X-ray spectroscopy have shown that an increase in the concentration of sulphate anions leads to a decrease in the concentration of higher order oligomers. This trend continues until the sulphate concentration reaches 2.0 M, after which the speciation of Zr and Hf is dominated by monomeric sulphate species (Hu et al., 2013; Kalaji et al, 2014). Fluoride nuclear magnetic resonance studies of aqueous TiF4 solutions indicated that only monomeric species exist in solution; where TiF4 is the dominant species (Serre et al., 2002). Furthermore, gas-phase computational chemistry studies predicted that dimerization of ZrF4 is energetically unfavourable (Voit et al., 2000).
In this study, computational chemistry was used to make predictions regarding the aqueous speciation of ZrF4 and HfF4. Specifically, density functional theory was used to predict three things: (1) how many aqua ligands associate to ZrF4 and HfF4 in aqueous solutions, (2) the aqueous Gibbs formation energies of ZrF4 and HfF4, and (3) the Gibbs reaction energies for the stepwise hydrolysis and fluoride dissociation reactions of ZrF4 and HfF4. Knowing how many aqua ligands are associated to ZrF4 and HfF4 is vital to accurately predict Gibbs reaction energies involving these complexes. Furthermore, such knowledge also allows rationalization of how many extractants can bind to these two complexes during SX reactions, since extractants often replace aqua ligands when binding to a metal. Since experimental data for the aqueous Gibbs formation energies of ZrF4 and HfF4 is reported in the literature (Brown et al., 2005), predicting these values and comparing them to the literature values allows assessment of the accuracy of the computational chemistry method that was used in this study.
After assessing the accuracy of the computational approach that was used, the dissociation constants, K, which correspond to various metal complexes in equilibrium (House, 2013), were predicted. Dissociation constants were determined by the following thermodynamic relation:

where pK = -logK, ΔG is the change in Gibbs energy corresponding to a given dissociation reaction, R is the ideal gas constant, and T temperature (Schroeder, 2000).
Method
The computational predictions presented in this paper were done using the DMol3code as implemented in Biovia's Materials Studio computational chemistry software package (Delley, 1990, 2000). All calculations were done within the framework of density functional theory using either the PBE (Perdew, Burke, and Ernzerhof, 1996, or B3LYP (Becke, 1993) functionals. The DMol3 code uses numerical basis sets. The basis sets used in this study, which are referred to as DNP basis sets in the DMol3 code, involve representing each valence orbital by two primitive functions plus a third polarized function. These DNP basis sets also treat the core electrons of Zr and Hf with relativistic core potentials. Since the functionals used in this study do not account for dispersion interactions, dispersion interaction corrections were added by using the Grimme scheme (Grimme, 2006). For all structures on which geometrical optimization calculations were done, vibrational frequency calculations were also used to confirm that these geometrical optimization calculations resulted in structures that represent potential energy minima.
Results and discussion
The first step in this work was to predict how many aqua ligands coordinate to ZrF4 and HfF4 in aqueous solutions. To do this, a ZrF4 molecules was subjected to geometry optimization calculations, for 200 optimization steps, in the presence of 26 water molecules. The 26 water molecules were arranged in a random fashion so that they were evenly distributed around the ZrF4, as shown in Figure 1. The choice to use 26 water molecules was based on the judgement that 26 water molecules would sufficiently surround the ZrF4 molecule in such a way that the solvation environment of ZrF4 would be adequately described. This procedure was repeated for ZrF3+, ZrF22+, ZrF3+, and Zr4+, where each Zr complex was surrounded by water molecules in a unique orientation.
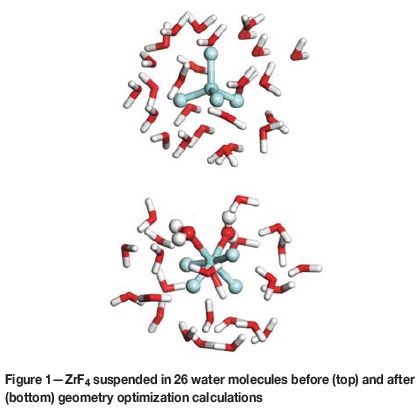
The geometry optimization calculations that were done on the various Zr complexes predicted that for each complex the Zr atom would coordinate to some of the surrounding water molecules. Therefore, for each complex, the Zr atom was predicted to have, together with the number of initial fluoride ligands, a total of six ligands coordinated to it. For instance, ZrF4 coordinated with two water molecules, ZrF3+ with three water molecules, etc. Figure 1 shows the coordination of two water molecules to ZrF4. Since each Zr structure was predicted to have six ligands, it was assumed that six ligands represent a stable coordination number for Zr fluorides.
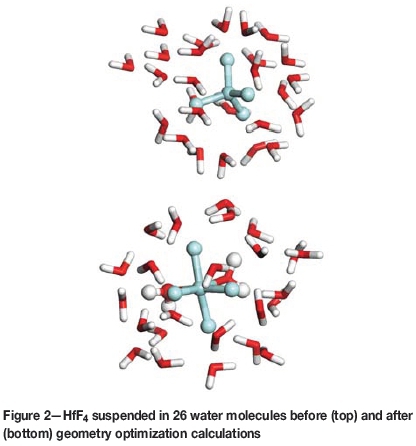
The procedure outlined above was repeated for HfF4, HfF3+, HfF22+, HfF3+, and Hf4+, where each Hf complex was surrounded by 26 water molecules and subjected to geometry optimization calculations (see Figure 2). Similar to the Zr complexes, all the Hf complexes were predicted to have structures where six ligands where coordinated to the Hf metal. Therefore, the assumption that six ligands represent a stable coordination number was also made for Hf fluorides.
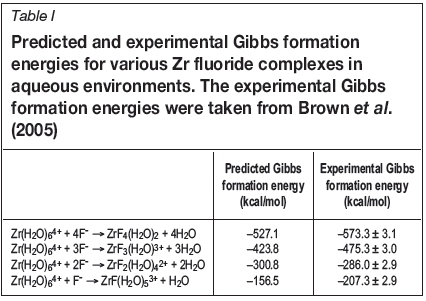
Once it was established, within the context of the predictions that were made in this work, that Zr and Hf fluorides coordinate with aqua ligands, each forming six-coordinate complexes, the Gibbs formation energies for Zr fluoride complexes in aqueous solutions were predicted. As stated before, the aim of predicting these Gibbs formation energies is to allow assessment of the accuracy of the computational method used in this work. The predicted Gibbs formation energies are given in Table I. To be clear, these
Gibbs formation energies refer to the formation of Zr fluoride complexes in aqueous environments. For instance, the formation of ZrF4 in aqueous environments is represented by the equation
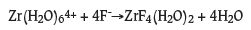
where six water ligands are coordinated to the Zr4+ ion and two aqua ligands are coordinated to ZrF4; representing a hydrated Zr4+ ion and hydrated a ZrF4 molecule. Therefore, these predicted Gibbs formation energies do not include the Gibbs formation energy that would be associated with the formation of water molecules.
When comparing the predicted Gibbs formation energies to the corresponding experimental values that were taken from literature, it is clear that the computational chemistry predictions presented in this paper are in qualitative
agreement with the experimental results. However, since these predictions are not in quantitative agreement with the experimental values, the results in this paper should only be used to suggest trends in the aqueous speciation of ZrF4 and HfF4.
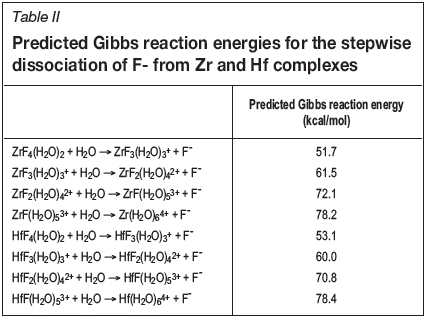
The predictions for the stepwise dissociation of fluoride ligands from Zr and Hf complexes are shown in Table II. Since these predicted Gibbs reaction energies are so large, it follows that the Zr-F and Hf-F bonds were predicted to be very stable. This is not surprising, given that fluoride is a very hard Lewis acid, while Zr(IV) and Hf(IV) are very hard Lewis bases. Furthermore, it is also predicted that ZrF4 and HfF4 are by far the most stable complexes of all the Zr and Hf fluorides modelled in this work. Therefore, the stepwise hydrolysis of Zr and Hf fluorides was modelled under the assumption that Zr and Hf are always present as ZrF4 and HfF4.
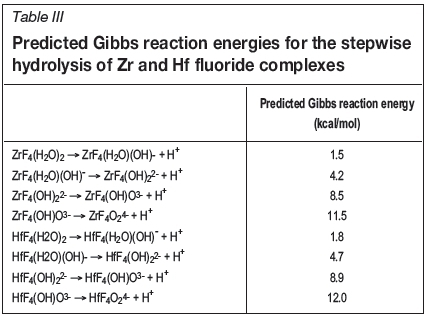
The Gibbs reaction energies for the stepwise hydrolysis of ZrF4 and HfF4 are shown in Table III. Figures 3 and 4 show the distribution of species diagrams for zirconium and hafnium tetrafluoride species, respectively. It is not surprising that the prediction reaction energies for the hydrolysis and the distribution of species are very similar, since Zr and Hf have very similar chemical properties.
A noteworthy difference between Zr and Hf is that while for Zr the first hydrolysis product is predicted to appear at a pH of 0.0, the same hydrolysis product for Hf is predict to appear at a pH of 2.0. Again, these results should be understood as only a prediction of trends. Therefore, these results predict that at very low pH values, extractants that bind to anionic species should be selective towards Zr.
Another important difference between Zr and Hf is that for Hf the first hydrolysis product is predicted to be present in low concentrations, never making up more that 40% of the species present in the Hf aqueous solution.
At a pH of 4.0, the aqueous solutions of both Zr and Hf are predicted to be dominated by dihydroxo complexes. Finally, at a pH of 6.5, both solutions are predicted to be dominated by the third hydrolysis product, namely tetrafluorohydroxooxo complexes of Zr and Hf. Taken together, these trends suggest that for higher pH values, both solutions show similar speciation characteristics.
Conclusion
In this work, density functional theory was used to make predictions about the aqueous speciation of Zr and Hf fluorides. It was predicted that if ZrF4or HfF4is dissolved in an aqueous solution, none of the fluoride ligands would dissociate. However, the degree to which aqueous tetrafluoride zirconium or hafnium complexes hydrolyse depends on the pH of the solution. It was predicted that above pH values of 0.0, hydrolysed species exist for Zr, and for a pH above 2.0 hydrolysed species exist for Hf.
It should be noted that the modelling presented in this paper was done in such a way that the possibility of two complexes reacting was not accounted for. The predictions made here therefore pertain only to the limiting case of the speciation of monomeric species. Nevertheless, inferences can be made about the possibilities of polymerization. Since ZrF4 was predicted to hydrolyse above a pH value of 0.0, while HfF4was predicted to hydrolyse above a pH of 2.0, both metals can polymerize via hydroxide or oxide bridges at pH values above 2.0. However, given that the rate of ageing is inversely proportional to the concentration of hydrolysed species, unaged solutions of pH values above 2.0 are predicted to be dominated by monomeric species.
Given that the fluorine ligands do not dissociate from the central atom in aqueous environments, it is predicted that the species in solution will be either negatively charged or neutral. The charge of the dominant species depends on the pH; for instance, at pH values below 1.0, the dominant species is neutrally charged.
Given the predictions regarding the ageing of the solution and the charge-dominant species, it is recommended that SX of ZrF4 and HfF4 be done at a pH of about 1.0 using unaged aqueous phases.
Acknowledgements
The authors would like to thank the South African Nuclear Energy Corporation SOC Limited (Necsa) and the New Metals Development Network (NMDN) of the Advanced Metals Initiative (AMI) of the Department of Science and Technology (DST) for their financial support. The authors would also like to thank the South African Center for High Performance Computing (CHPC) for the use of their computational resources.
References
Banda, R. and Lee, M.S. 2015. Solvent extraction for the separation of Zr and Hf from aqueous solutions. Separation and Purification Reviews, vol. 44, no. 3. pp. 199-215. [ Links ]
Becke, A.D. 1993. Density-functional thermochemistry. III. The role of exact exchange. Journal of Chemical Physics, vol. 98, no. 7. pp. 5648-5652. [ Links ]
Blumenthal, W.B. 1958. The Chemical Behavior of Zirconium. 1st edn. Literary Licensing, New York. [ Links ]
Brown, P.L., Mompean, F.J., Perrone, J., and Illemassène, M. 2005. Chemical Thermodynamics of Zirconium. 1st edn. Elsevier, Amsterdam. [ Links ]
Delley, B. 1990. An all-electron numerical method for solving the local density functional for polyatomic molecules. Journal of Chemical Physics, vol. 92. pp. 508-517. [ Links ]
Delley, B. 2000. From molecules to solids with the DMol3 approach. Journal of Chemical Physics, vol. 113. pp. 7756-7764. [ Links ]
Grimme, S. 2006. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. Journal of Computational Chemistry, vol. 27, no. 15. pp. 1787-1799. [ Links ]
Hagfeldt, C., Kessler, V., and Persson, I. 2004. Structure of the hydrated, hydrolysed and solvated zirconium(IV) and hafnium(IV) ions in water and aprotic oxygen donor solvents. A crystallographic, EXAFS spectroscopic and large angle X-ray scattering study. Dalton Transactions, vol. 14. pp. 2142-2151. [ Links ]
House, J.E. 2013. Inorganic Chemistry. 2nd edn. Academic Press, Oxford, UK. [ Links ]
Hu, Y.J., Knope, K., Skanthakumar, S., Kanatzidis, M., Mitchell, J. and Soderholm, L. 2013. Understanding the role of aqueous solution speciation and its application to the directed syntheses of complex oxidic Zr chlorides and sulfates. Journal of the American Chemical Society, vol. 135. pp.14240-14248. [ Links ]
Kalaji, A., Skanthakumar, S., Kanatzidis, M., Mitchell, J., and Soderholm, L. 2014. Changing hafnium speciation in aqueous sulfate solutions: A high-energy X-ray scattering study. Inorganic Chemistry, vol. 53. pp. 6321-6328. [ Links ]
Perdew, J.P., Burke, K., and Ernzerhof, M., 1996. Generalized gradient approximation made simple. Physical Review Letters, vol. 77, no. 18. pp. 3865-3868. [ Links ]
Schroeder, D.V. 2000. An Introduction to Thermal Physics. 1st edn. Pearson, Edinburgh, UK. [ Links ]
Serre, C.; Corbière, T.; Lorentz, C.; Taulelle, F. and Férey, G. 2002. Hydrothermal synthesis of nanoporous metalofluorophosphates. 1. Precursor solutions of titanium fluoride and fluorophosphate in water, a 19F and 31P NMR study. Chemistry of Materials, vol. 14. pp. 4939-4947. [ Links ]
Voit, E., Voit, A., Gerasimenko, A., and Sergienko, V. 2000. Quantum chemical study of model fluorozirconate clusters. Journal of Structural Chemistry, vol. 41. pp. 41-47. [ Links ] ♦
Paper received Sept. 2016
revised paper received Mar. 2018


{kind=link}
{kind=link}