










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkJournal of the Southern African Institute of Mining and Metallurgy
versão On-line ISSN 2411-9717
versão impressa ISSN 2225-6253
J. S. Afr. Inst. Min. Metall. vol.117 no.8 Johannesburg Ago. 2017
http://dx.doi.org/10.17159/2411-9717/2017/v117n8a9
HYDROMETALLURGY CONFERENCE 2016
Recovery of uranium from nuclear conversion plant waste
M. PotgieterI, II; J.C. BarryI; D.J. van der WesthuizenI; H.M. KriegI
IUranium Chemistry Group, Plasma Technology, Applied Chemistry, Nuclear Energy Corporation of South Africa (Necsa), South Africa
IIMembrane Technology Group, Chemical Resource Beneficiation (CRB), North-West University (NWU), South Africa
SYNOPSIS
The ammonium diuranate (ADU) conversion process that was operated at the Nuclear Energy Corporation of South Africa (Necsa) in the past generated a significant amount of waste containing high concentrations of uranium, which can be re-used if the uranium can be recovered in a useful form. To attain this objective, the composition of the waste material and the amounts of impurities present were determined, followed by an investigation into various methods of uranium dissolution. For dissolution, water as well as different acid types and concentrations were investigated, and the efficiency of each method determined in terms of the uranium recovery as well as the extent of impurities extraction. It was found that the waste material was soluble in HNO3, H2SO4, and HCl as well as water, with a maximum uranium extraction of 98% achieved in 3 M HNO3 in 1 hour at a temperature of 80°C without the addition of an oxidizing agent. The thorium impurity content in relation to uranium was reduced from 8.4% to less than 1% with all acids investigated, as well as water. The most significant reduction in the total impurity content, from 24.3% to 10.8 %, was observed when using water, although this did not result in the highest uranium extraction.
Keywords: uranium, nuclear waste, conversion process, dissolution, purification, hydrogen peroxide.
Introduction
The ammonium diuranate (ADU) conversion process operated in South Africa from the 1970s was based on the direct conversion of ADU obtained from different mines (Ponelis, Slabber, and Zimmer, 1986; Ponelis, 1989). This approach differed from those of other countries operating conversion processes at that time in that the feed was not of nuclear grade. Since the ADU was sourced from various South African mines, the composition of the feed to the conversion plant at the Atomic Energy Corporation (AEC) varied. In addition, tail-end distillation of the uranium hexafluoride (UF6) product was used, and as a result of the variability in the ADU a considerable proportion of unwanted elements or impurities, such as sodium (Na), potassium (K), and calcium (Ca), was present during the conversion process (Ponelis, Slabber, and Zimmer, 1986; Ponelis, 1989).
Basically, the ADU conversion process entailed the conversion of ADU to uranium tetrafluoride (UF4), which was then converted to uranium hexafluoride (UF6) in a fluorine flame reactor (Ponelis, 1989). The final product, thermally stable UF6gas, was then filtered to remove any foreign particles. Thereafter, the UF6was frozen out and distilled before being fed to the enrichment plant. Due to the incomplete conversion of UF4 to UF6in the fluorine flame reactor, a significant amount of solid waste rich in uranium was formed, which accumulated at the bottom of the reactor. The incomplete conversion could have been caused by various factors, including sintering due to the low melting point of UF4, inhomogeneous feed compositions, and the presence of various impurities. A study to determine the effect of alkali metal impurities on the conversion of UF4to UF6showed that the presence of these impurities had a significant effect on the sintering of UF4 (Ponelis, 1989).
Owing to the high uranium content, the unreacted material is a nuclear liability and needs to be processed to recover a product that can be re-used in possible future conversion activities while reducing the amount of waste that is currently stored in drums. A search of the literature was undertaken to determine whether similar waste exists elsewhere, and how it is handled. Sasahira et al. (2007) mention an 'ash' that was formed during fluorination of UO2 to UF6, while Ohashi, Murashita, and Nomura (2014) extracted uranium from UF4 residue and NaF adsorbents originating from conversion activities. It thus became clear that this waste material is unique, in terms of its intrinsic variety in composition as well as the amounts and types of impurities present.
In view of the solvent extraction (SX) process that is used to purify uranium (Kumar et al., 2011), the waste material in its current solid form will have to be dissolved to obtain an aqueous solution containing the desired uranium. Furthermore, in order to extract uranium successfully using SX, the fluoride content should be low since fluoride is co-extracted by tributyl phosphate (TBP) (Volk, Vakhrushin, and Mamaev, 2000; Coleman, 1966).
It can be assumed that a significant amount of the contained uranium is in the UF4form, since the waste material was generated in the UF4to UF6conversion step in the process, and unreacted material would therefore consist of UF4. For the envisioned SX process, provision should be made for the oxidation of insoluble U(IV) to soluble U(VI) during dissolution, possibly by means of the addition of an oxidizing agent. UF4 can be completely solubilized by direct treatment with concentrated nitric acid (HNO3) (Floreancig, 1983, Ohashi, Murashita, and Nomura, 2014). Luk'yanchev and Nikolaev (1963) studied the dissolution of UF4 in sulphuric (H2SO4) and hydrochloric (HCl) acid, and found that the solubility of UF4 increases with increasing HCl concentration, and reaches a maximum in H2SO4at approximately 3 M. Ohashi, Murashita, and Nomura (2014) used an oxidizing agent (H2O2) during dissolution of UF4 in H2SO4and HCl as part of a study on the extraction of uranium from fluorine-containing waste. They found that UF4 could be completely dissolved in 1.25 M HCl or 0.3 M H2SO4.
The aim of this investigation was, therefore, to dissolve the current waste material and to obtain the highest possible recovery of uranium in solution. Since no reference to similar waste material could be found, and a low solubility was expected due to its sintered nature, the waste material was milled prior to dissolution. The dissolution efficiency was investigated using water as well as different acids (HNO3, H2SO4, and HCl) with and without the addition of H2O2as an oxidizing agent. Water was included as a washing step prior to dissolution to determine its suitability for removing any water-soluble impurities. Since the waste material is known to contain high levels of fluoride (approx. 20%), different methods will be investigated for removing fluoride subsequent to dissolution but prior to SX, but this falls beyond the scope of the current study. In this paper the focus is mainly on cations in the waste material with concentrations above 0.5 mass%.
Experimental
Composition of waste material
The composition of the waste material was determined, focusing on the uranium (U) content and other major constituents that may ultimately influence the SX-based uranium recovery process. A sample of the waste material was dissolved in nitric acid (65 wt% HNO3) in a 1:1 water to acid ratio. The solution was evaporated to dryness and the residue was re-dissolved in HNO3at the same concentration to achieve complete dissolution, followed by analysis using inductively coupled plasma-optical emission spectrometry (ICP-OES).
Dissolution tests
Nitric acid (HNO3, 65 wt%), sulphuric acid (H2SO4, 98 wt%), and hydrochloric acid (HCl, 32 wt%) were obtained from Merck. The dissolution tests were conducted using HNO3 at concentrations of 3 M and 1 M, H2SO4 at 2.5 M and 1.25 M, and HCl at 3 M and 1 M, as well as ultrapure water. Experiments were conducted in the absence and presence of 0.4 mL H2O2 (30 wt%, from Merck) as an oxidizing agent to aid the oxidation of insoluble U(IV) to soluble U(VI). For the solubility measurements, a 1 g sample of the waste material was added to a 100 mL acid solution and heated to 80°C on a hot plate. The temperature was chosen to be high enough to ensure fast reaction kinetics, but low enough to ensure that the solution did not boil, which would cause unwanted loss of solution as well as the evolution of noxious fumes. Each dissolution experiment was continued for 1 hour with continuous stirring using a stirrer bar, to ensure sufficient time for the reaction to proceed, after which the mixture was left to cool to room temperature. The mixture was then centrifuged and decanted to separate the solution from the residue. The residue was washed with distilled water, dried in an oven at 100°C, and weighed. A sample of each solution and residue was analysed using ICP-OES.
Using the acid with which the highest uranium recovery was attained, a more comprehensive study was subsequently undertaken in which additional acid concentrations were investigated under identical experimental conditions to the screening investigation described above.
Results and discussion
Composition of waste material
A rough estimate of the composition of the waste material was determined (data not shown) by X-ray fluorescence (XRF). Based on this data, the cations present at concentrations greater than 0.5% by mass were determined using ICP-OES. The results are shown in Table I.
Since uranium will be recovered by means of SX, lowering the amount of impurities that may have an influence on the SX process would be an added benefit. From Table I it is clear that Fe (28.0 mg/g), Th (44.5 mg/g), and Ca (50.0 mg/g) are the main impurity elements in the waste material. Currently, there are no clear specifications for the final product material that would result from recovery of uranium. However, it is envisaged that the product would be re-used in a nuclear conversion plant. Therefore, the ASTM standard for uranium ore concentrate (UOC) can be used to specify the maximum allowed levels of impurities of the final product. These are 1% for both calcium and iron and 2.5% for thorium (ASTM, 2013). The focus of the current study was to dissolve the waste material as a preparatory step for future purification by SX, and lowering the levels of these impurities would be a benefit if it can be achieved during this step. To facilitate the discussion on impurities, the content of each impurity was expressed as a percentage of the uranium according to the relationship:
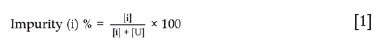
where i and U refer to the impurity and uranium respectively. The values are presented in Table II.

As mentioned previously, it can be assumed that most of the impurities originate from the feed material to the conversion process. Accordingly, the high thorium content resulted from the non-volatility of the formed ThF4, which caused it to settle at to the bottom of the flame reactor together with the rest of the waste material not converted to UF6. Since the residue was re-fed to the flame reactor to reduce the amount of waste, thorium as well as other impurities were further concentrated in relation to uranium.
Dissolution
Screening of lixiviants
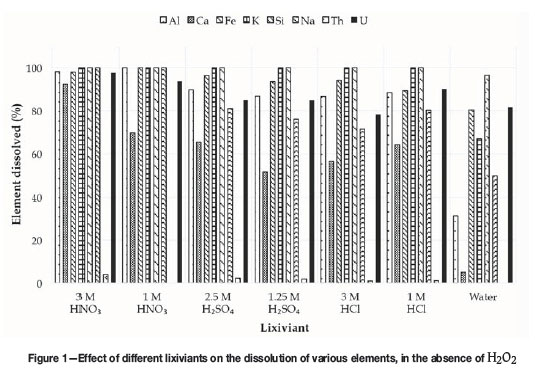
To identify the most promising lixiviant for the extraction of uranium from the waste material, a series of screening experiments was conducted using nitric, sulphuric, and hydrochloric acids at two concentrations, with and without addition of H2O2. Another objective was to determine whether the more water-soluble impurities could be removed during this step, even if uranium dissolution was not achieved. The results for the screening experiments in the absence of H2O2are presented in Figure 1. The percentage dissolution of each element was calculated using the following relationship:
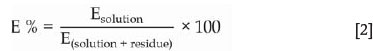
where E represents the specific element investigated.
It can be seen that the uranium dissolution was above 78% for all the acids, and 82% for water, with the highest uranium recovery (98%) observed in 3 M HNO3. It is noteworthy that the thorium concentration in solution remained below 5% in all instances. The solubility of calcium, however, was significantly different in in different acids, and very limited in water. Although the calcium content in the waste material could be due to a mixture of various compounds, the behaviour observed could be explained by the presence of calcium sulphate dihydrate (CaSO4.2H2O), the solubility of which is 0.222 g/100 mL in hot water and 0.241 g/ 100 mL in cold water (CRC, 1976). Other calcium compounds show either very high or very low solubilities in water.
The solubility of CaSO4.2H2O increases with an increase in acid concentration up to a maximum value (3.5 to 4.5 M for HNO3, 2.5 to 3 M for HCl, and 1.5 M for H2SO4), and then decreases if the acid concentration is increased further (Vershkova et al., 2003; Cameron and Breazeale, 1903; Ling and Demopoulos, 2004; Li and Demopoulos, 2005). For H2SO4, the initial increase in solubility of CaSO4.2H2O has been attributed to the influence of the second dissociation constant of H2SO4, due to the higher solubility in the presence of HSO4- as well as to the increase in ionic strength in the presence of more H2SO4 (Marshall and Jones, 1966). The decrease in solubility at higher concentrations is believed to be due to a combination of effects which include changes in activity coefficient as well as salting-out due to the common-ion effect (Ling and Demopoulos, 2004; Calmanovici et al., 1993). The increasing and decreasing solubility in HNO3 was not clearly explained by Vershkova et al. (2003); however, Zhang et al. (2011) observed a decrease in the solubility of CaSO4.2H2O in the order HNO3 > H3PO4 > H2SO4 > Ca(NO3)2, also without offering an explanation, but which agrees with the results we obtained. A possible explanation for the behaviour in HCl was given by Li and Demopoulos (2005), who ascribed the initial increase in solubility to the presence of HSO4- formed during dissolution of CaSO4.2H2O, and the decrease at higher HCl concentrations to the influence of ion activity coefficients.
While the values in Figure 1 cannot be compared directly with those from the literature, due to the complexity of this waste material, the above does explain the higher calcium extractions at higher HNO3and H2SO4concentrations. The lower calcium extraction at higher HCl concentration may indicate that the maximum CaSO4.2H2O solubility was at a HCl concentration below 3 M.
The purities of the final uranium materials (calculated using Equation [1]) with regard to the three most prevalent impurities (Ca, Fe, and Th) are given in Table III. It is clear that both calcium (in acid) as well as iron (all acids and water) are nearly completely extracted during dissolution (see Table II for original concentrations). Some concentrations are higher than the original concentrations since the change in the amount of uranium extracted (<100%) was used to calculate the percentage impurity.
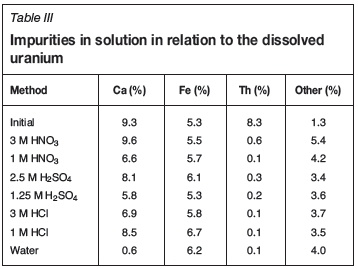
As mentioned above, the calcium level was significantly lowered from 9.3 to 0.6% by dissolution in water. The amount of iron was higher in all instances, with smaller differences from the initial value in instances where uranium recovery was higher (e.g. 3 M HNO3). The reason for this is that iron was dissolved more completely compared to the other impurities (thorium was very insoluble, and calcium solubility depended on acid type and concentration). Therefore, since the impurity values are calculated in terms of the total amount of uranium present, the values for iron as an impurity would also be higher, especially if the uranium recovery is lower. A more in-depth discussion on the behaviour of iron follows in the next section. The amount of thorium was lowered so significantly in all instances that all lixiviants are suitable for the separation of uranium and thorium during the dissolution step.
Influence of H2O2
The influence of H2O2 on the recovery of uranium in water and different acids at various concentrations, as well as on the purity of the final uranium solution, was determined. H2O2 acts as an oxidizing agent to facilitate the oxidation of the insoluble U(IV) to more soluble U(VI). The influence of H2O2 on the amount of impurities extracted, as well as the uranium recoveries for the different lixiviants, is presented in Figure 2.
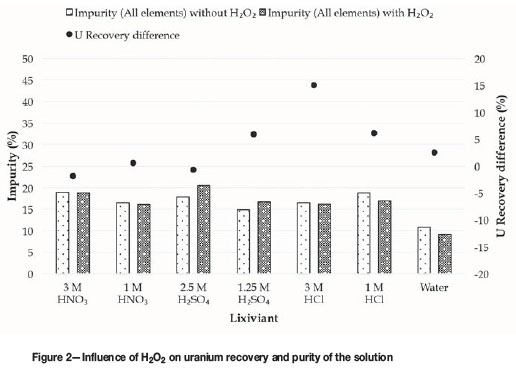
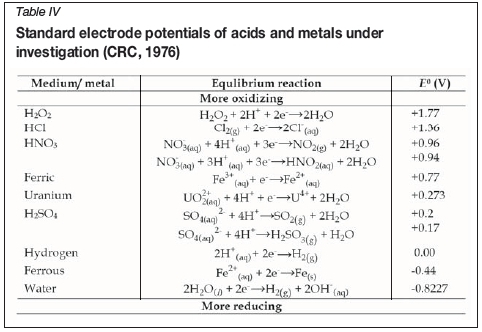
The secondary y-axis gives the difference in uranium recoveries in the absence and presence of H2O2. Therefore, a large positive value indicates a large increase in uranium recovery when H2O2 is added and a value of zero, no difference. The influence of H2O2 on uranium recovery was greatest when using 3 M HCl. In contrast, the influence of H2O2 was negligible when using HNO3and 2.5 M H2SO4. This can be explained in terms of the oxidizing ability of these acids. The standard electrode potentials (Table IV) show that HNO3 is the stronger oxidizing agent, whereas the oxidizing ability of H2SO4, increases at higher acid concentrations, hence the diminished influence of H2O2in the more concentrated H2SO4solution. However, the influence of H2O2, which was high with 3 M HCl, decreased again at the lower HCl concentration, and was low in water.
The bar chart (primary y-axis) in Figure 2 shows the effect of H2O2 on the impurity level of the final uranium solution, taking into account all elements and using Equation [1]. For the percentage impurity extraction in the absence of H2O2, the combined totals of the values presented in Table III were used. It is clear that the presence of H2O2had little influence on extraction of impurities. The purity of the solution increased slightly for HNO3, HCl, and water while decreasing slightly for H2SO4in the presence of H2O2.
As regards the three most abundant impurities (Ca, Fe, and Th), most of the thorium had already been removed in the absence of H2O2 (Table IV), and thorium removal was not further improved by adding H2O2. The effect of H2O2 on the dissolution of calcium and iron is shown in Figure 3.
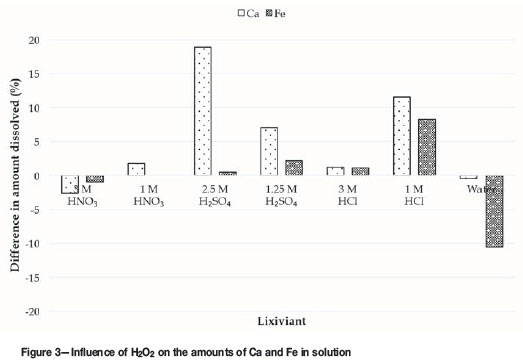
The reason for the decrease in solution purity for H2SO4 in the presence of H2O2 (Figure 2) becomes clear from Figure 3, which shows higher amounts of calcium and, in certain instances, iron dissolved. The increase in calcium dissolution may be due to a reaction between calcium and H2O2that results in the formation of a more acid-soluble calcium species such as calcium peroxide (CaO2) (Tsentsiper and Vasil'eva, 1967). At present, it is not viewed as critical to the uranium recovery aspect of this study for this aspect to be investigated further. Furthermore, a decrease in the amount of iron dissolved in water was observed when H2O2 is added.
The influence of H2O2 on iron is unexpected since the purpose of H2O2 addition was to interact with U(IV). The influence of H2O2 on uranium recovery in water (Figure 2) was also surprisingly low, since water is not a particularly oxidizing environment (see Table IV) and it was expected that H2O2 would increase uranium recovery in water.
The observations thus far suggest that the oxidizing ability of each lixiviant, the presence of H2O2, and the iron present in the waste material influence each other, thereby influencing the recovery of both uranium and impurities. Fe3+ is known to oxidize insoluble U(IV) to soluble U(VI) (Ervanne, 2004; Venter and Boylett, 2009) while being reduced to Fe2+ according to the following simplified equation:
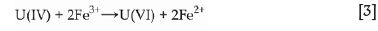
Since iron is present in the waste material in significant amounts, it may be possible that the oxidative dissolution of uranium is facilitated by Fe3+ and not directly by H2O2 as initially thought. In the presence of a strongly oxidizing acid such as HNO3, which is a stronger oxidizer than Fe3+ (Table IV), the oxidation of U(IV) is facilitated mostly by the acid itself and the effect of iron is masked. The H2O2 then rather acts as an oxidizing agent to regenerate the formed Fe2+ back to Fe3+, which is then again available to oxidize U(IV), as also described by Venter and Boylett (2009) for uranium leaching. The reaction of Fe2+ with H2O2in the presence of acid can be represented by the following equation:
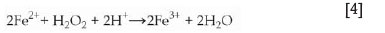
The higher uranium recovery in the presence of H2O2 with the lower concentration of H2SO4 can be explained by the lower oxidizing capability of H2SO4 at lower concentrations and its lower oxidizing capability compared with Fe3+, as seen in Table IV. Therefore, the Fe2+ formed during oxidation of U(IV) can be re-oxidized by H2O2to Fe3+, which will in turn enhance the uranium recovery. A similar effect was observed in 3 M HCl and since this is the least oxidizing environment, the addition of H2O2yielded the greatest enhancement of uranium recovery. The decrease in iron dissolution in water when H2O2is added may indicate the formation of an insoluble iron species, the nature of which is unknown and which would have to be further investigated.
Influence of HNO3concentration
In the initial tests the highest uranium recovery was achieved in HNO3. In addition, when using HNO3 systems, the addition of H2O2did not improve uranium recovery, while impurities such as iron had less of an influence on uranium recovery. To further optimize the dissolution, higher concentrations of HNO3 were used to confirm the observations made during the screening, where only two values in the lower concentration range were included. For this purpose the effect of HNO3 concentrations from 1 M to 12 M on the uranium recovery, as well as on the purity of the final solution, was investigated. The values for all elements dissolved in the different HNO3 concentrations are presented in Figure 4, calculated similarly as in Figure 1.
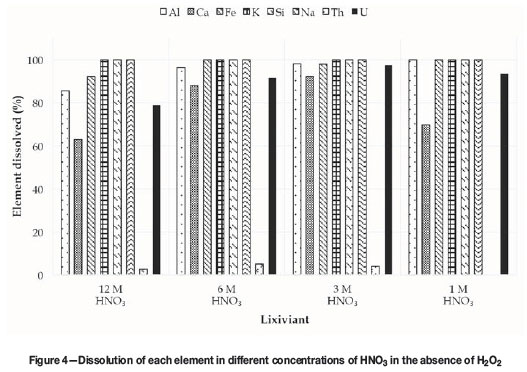
The recovery of uranium in HNO3 initially increased with increasing acid concentration, reaching a maximum of 98% at 3 M before declining to 78.75% in 12 M HNO3. Currently, there is no explanation for this decrease in recovery at higher HNO3 concentrations, and further investigation is needed.
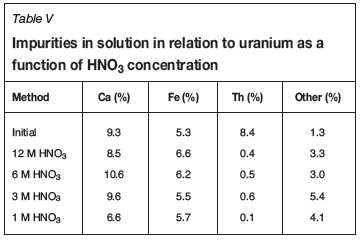
Calcium dissolution increased with increasing HNO3 concentration, from 69.83% at 1 M to 92.3% at 3 M, followed by a slight decrease to 88.05% at 6 M and then decreasing significantly to 63.03% at 12 M HNO3. These observations again agree with results in the literature in terms of the solubility of CaSO4.2H2O in HNO3, where an increase is observed with increasing acid concentration up to a certain point, followed by a decrease at higher concentrations (Vershkova et al, 2003). The amount of Ca, Fe, Th, and other impurities dissolved in relation to the amount of uranium dissolved was calculated using Equation [1], and the results are presented in Table V. Again, the thorium impurity was lowered from 8.4% to < 1% irrespective of the HNO3 concentration. The amount of iron increased in all HNO3 concentrations, with the lowest value at 3 M. For calcium, the lowest impurity levels were attained at 1 M and at 12 M, with the highest impurities at 6 M. The lowest total impurity level was obtained at 1 M HNO3.
Influence of H2O2 in HNO3
The results obtained when studying the influence of H2O2in HNO3 on uranium recovery and the purity of the final solution can be seen in Figure 5. The values of the bar chart (primary y-axis) represent the percentage impurity of the final solution calculated from all elements. The secondary y-axis shows the difference between the uranium recoveries in the absence and presence of H2O2.
These results confirm the observations from the screening tests, in that H2O2has limited influence on uranium recovery in an HNO3 environment and an insignificant influence on the purity of the final solution. Therefore, HNO3 would be an ideal choice for dissolution of the waste material, after which further purification will be conducted.
Conclusions
The nuclear conversion plant waste material is soluble in different acids. Different uranium recoveries were obtained at different acid concentrations, with the highest recovery (98%) being obtained in 3 M HNO3. Furthermore, for 3 M HNO3, it was not necessary to add an oxidizing agent to increase the uranium recovery or to decrease impurity levels. An advantage of all lixiviants studied is that thorium was largely removed from the waste material, decreasing from 8.4% to less than 1% in relation to the extracted uranium. The waste material was also leachable in water, with 82% uranium recovery combined with a significant reduction in the total amount of impurities in relation to uranium (from 24.3% to 10.8%).
The influence of H2O2 on uranium recovery was found to be greatest in less oxidizing environments, especially in 3 M HCl. This is probably due to the role of Fe3+ as an oxidizing agent for the oxidative dissolution of U(IV), Fe3+ being in turn reduced to Fe2+. The H2O2 then acts to regenerate Fe2+ to Fe3+, which is once again available for oxidation of U(IV). In view of the significant amounts of fluoride present in the waste material, future work to will investigate suitable methods for fluoride removal.
Due to the environmental liability around the conversion plant waste material, successful dissolution thereof opens up possibilities for re-use of the material once the uranium has been purified. Not only will the amount of waste in storage be reduced, but a valuable product will be obtained that may possibly be used as feed material to a conversion process.
Acknowledgements
We would like to thank the National Research Foundation (NRF), the Nuclear Energy Corporation of South Africa (Necsa), and the Chemical Resource Beneficiation (CRB) group for funding that made this work possible. We would also like to acknowledge Mr L. Nkadimeng for help with experimental work.
References
ASTM. 2013. Standard specification for uranium ore concentrate. 3. ASTM International, West Conshohocken, PA. [ Links ]
Calmanovici, C.E., Gabas, N., and Laguérie, C. 1993. Solubility measurements for calcium sulfate dihydrate in acid solutions at 20, 50 and 70°C. Journal of Chemical Engineering and Data, vol. 38. pp. 534-536. [ Links ]
Cameron, F.K. and Breazeale, J.F. 1903. Solubility of calcium sulphate in aqueous solutions of sulphuric acid. Journal of Physical Chemistry, vol. 7. pp. 271-577. [ Links ]
Coleman, C.F. 1966. Recovery of uranium and zirconium from aqueous fluoride solutions. US patent US3243257. [ Links ]
CRC. 1976. Handbook of Chemistry and Physics. CRC Press, Boca Raton, FL. [ Links ]
Ervanne, H. 2004. Detecting and minimizing interferences in uranium oxidation states during dissolution of solid phases. Journal of Radioanalytical and Nuclear Chemistry, vol. 260. pp. 249-253. [ Links ]
Floreancig, A. 1983. Method of dissolving impure uranium tetrafluoride. France patent application. [ Links ]
Kumar, J.R., Kim, J.-S., Lee, J.-Y., and Yoon, H.-S. 2011. A brief review on solvent extraction of uranium from acidic solutions. Separation and Purification Reviews, vol. 40. pp. 77-125. [ Links ]
Li, Z. and Demopoulos, G.P. 2005. Solubility of CaSO4 phases in aqueous HCl + CaCl2 solutions from 283 K to 353 K. Journal of Chemical and Engineering Data, vol. 50. pp. 1971-1982. [ Links ]
Ling, Y. and Demopoulos, G.P. 2004. Solubility of calcium sulfate hydrates in (0 to 3.5) mol.kg-1 sulfuric acid solutions at 100°C. Journal of Chemical and Engineering Data, vol. 49. pp. 1263-1268. [ Links ]
Luk'yanchev, Y. and Nikolaev, N.S. 1963. The solubility of uranium tetrafluoride in aqueous solutions of acids. Atomic Energy, vol. 15. pp. 1184-1187. [ Links ]
Marshall, W.L. and Jones, E.V. 1966. Second dissociation constant of sulfuric acid from 25 to 350° evaluated from solubilities of calcium sulfate in sulfuric acid solutions. Journal of Physical Chemistry, vol. 70. pp. 4028-4040. [ Links ]
Ohashi, Y., Murashita, S., and Nomura, M. 2014. Extraction of uranium from solid waste containing uranium and fluorine. Minerals Engineering, vol. 61. pp. 32-39. [ Links ]
Ponelis, A.A. 1989. Die invloed van alkali-metaalonsuiwerhede op die uraandioksiedhidrofluorineringsreaksie. PhD thesis, University of Pretoria. [ Links ]
Ponelis, A.A., Slabber, M.N., and Zimmer, C.H.E. 1986. Conversion of non-nuclear grade feedstock to UF4. Proceedings of Advances in Uranium Refining and Conversion. International Atomic Energy Agency, Vienna. pp. 111-139. [ Links ]
Sasahira, A., Kani, Y., Iino, K., Hoshino, K., and Kawamura, F. 2007. Development of FLUOREX process as a progressive LWR reprocessing system. Proceedings of the Global 2007 Conference on Advanced Nuclear Fuel Cycles and Systems, Boise, Idaho, 9-13 September 2007. American Nuclear Society, La Grange Park, IL [ Links ]
Tsentsiper, A.B. and Vasil'eva, R.P. 1967. The reaction of calcium hydroxide with hydrogen peroxide vapor. Bulletin of the Academy of Sciences of the USSR, Division of Chemical Science, vol. 16. pp. 2445-2447. [ Links ]
Venter, R. and Boylett, M. 2009. The evaluation of various oxidants used in acid leaching of uranium. Proceedings of Hydrometallurgy 2009. Southern African Institute of Mining and Metallurgy, Johannesburg. pp. 445-255. [ Links ]
Vershkova, Y.A., Tareeva, O.A., Ivlev, K.G.M., and Lokshin, E.P. 2003. Solubility of calcium sulfate dihydrate in nitric acid at 20°C. Russian Journal of Applied Chemistry, vol. 76. pp. 156-157. [ Links ]
Volk, V.l., Vakhrushin, A.Y., and Mamaev, S.L. 2000. Extraction of uranium and thorium with TBP from fluoride - nitric acid solutions. Journal of Radioanalytical and Nuclear Chemistry, vol. 246. pp. 697-702. [ Links ]
Zhang, Q.Y., Huang, X.H., Tang, H.B., and He, H. 2011. Electrochemical behavior of uranium(VI) in 1-butyl-3-methylimidazolium chloride. He-Huaxueyu Fangshe Huaxue (Journal of Nuclear and Radiochemistry), vol. 33. pp. 101-105. [ Links ]


{kind=link}
{kind=link}