










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkJournal of the Southern African Institute of Mining and Metallurgy
versión On-line ISSN 2411-9717
versión impresa ISSN 2225-6253
J. S. Afr. Inst. Min. Metall. vol.117 no.7 Johannesburg jul. 2017
http://dx.doi.org/10.17159/2411-9717/2017/v117n7a3
YOUNG PROFESSIONALS CONFERENCE
Accurate measurement of polarization potentials during electrodeposition of nickel metal from sulphate electrolytes
L. Schoeman; K.C. Sole
University of Pretoria, South Africa
SYNOPSIS
Extraction of nickel from either latente or sulphide ores involves various complex hydrometallurgical processes, the final step of which is often electrowinning to produce pure nickel metal. Methods to investigate, monitor, and control the electrowinning process are therefore of importance for producing nickel metal of desirable quality and purity. The significance of potential measurements during the electrowinning process is well established. Many potential measurement methods, however, are not sufficiently accurate or repeatable, and it is therefore often difficult to monitor and control the electrodeposition process accurately. The relationship between nucleation (deposition of the first nickel clusters to the cathode surface) and growth (as more and more nickel clusters deposit) during electrodeposition is paramount for controlling the formation and quality of the nickel metal electrodeposit. A need therefore still exists for accurate measurement of both nucleation and plating overpotentials during the early stages of electrodeposition. In this investigation, a polarization measurement technique was developed for nickel electrodeposition from sulphate electrolytes under typical commercial electrowinning conditions. This method is repeatable and accurate within approximately 10 mV, and enables the quick and concise monitoring of both nucleation and plating during nickel electrodeposition.
Keywords: nickel electrowinning, polarization potential measurement, sulphate electrolyte, galvanodynamic scan.
Introduction
Nickel electrodeposition reactions
In order to evaluate nickel electrodeposition, it is imperative to understand the reactions taking place in the electrolyte. Conditions should be conducive to promoting the main nickel deposition reaction while limiting unwanted or side reactions that could hinder or interfere with the main deposition reaction. The process of nickel electrodeposition from sulphate electrolyte involves the passing of a direct current between two electrodes immersed in a sufficiently conductive solution of nickel sulphate salt. Nickel cations are reduced to nickel metal at the cathode (Equation [1]) while oxidation of water at the anode (Equation [2]) releases the required electrons. The nickel ions consumed at the cathode are replenished from the nickel ions in the electrolyte. All standard potentials are given relative to the Ag/AgCl (saturated KCl) reference electrode (Holm and O'Keefe, 2000; Crundwell et al., 2011):
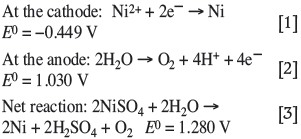
The reduction of species other than that of the sought metal should be avoided because this will reduce the current efficiency of the nickel metal plating process, and interfere with the nature and purity of the plated metal. In the case of nickel specifically, the nickel ions are thermodynamically more stable in aqueous solutions than hydrogen ions. Therefore, the reduction of hydrogen to hydrogen gas (Equation [4]) is thermodynamically more favourable and will occur simultaneously with the reduction of nickel cations (Holm and O'Keefe, 2000; Kittelty, 2002; Di Bari, 2010; Crundwell et al., 2011):
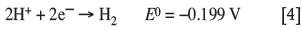
This reaction needs to be controlled or minimized in such a way that nickel cation reduction is optimally promoted. One such precaution to minimize the interference of hydrogen bubbles is used almost exclusively, in which the anode and cathode are separated by an ion-permeable membrane. The electrolyte ions can freely move through the membrane, but gas bubbles are trapped on either side where they are produced. Membranes, or cathode bags, are commonly made from a material such as polyethylene or polyester (Crundwell et al., 2011).
Another factor that needs to be kept in mind is that, although the stoichiometry and thermodynamics are accurately described by the reactions given above, the simultaneous transfer of more than one electron in an electrochemical process is unlikely to occur. There are many proposed mechanisms for the nickel reduction process, most of which assume that electrocrystal-lization occurs in two steps of electron transfer involving an intermediate adsorbed species (Equations [5] and [6]) (Aaboudi et al., 2001; Kittelty, 2002; Di Bari, 2010, Crundwell et al., 2011):
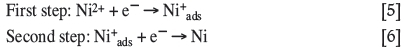
Even though there are many proposed ligands, the most common and simplest one to consider is the hydroxide ion that originates from the water present in the electrolyte solution. Hydroxides easily coordinate to the nickel ions according to the proposed reactions in Equations [7] to [10] (Munoz et al., 2003; Di Bari, 2010). The charge-transfer reaction responsible for the formation of the Ni(OH)ads complex (Equation [8]) is typically considered as the rate-determining step (Di Bari, 2010). The Ni(OH)ads complex is believed to act either as a catalyst for the reduction of the metal ion or as an intermediate that is consumed to form the reduced metal species, as shown in Equations [9] and [10] (Munoz et al., 2003; Di Bari, 2010):
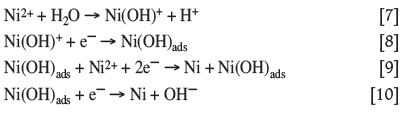
Effect ofconditions and parameters on nickel electrodeposition
In order to obtain good quality nickel electrodeposits with a fine-grained, uniform morphology, low strain, and minimal hydrogen pitting, frequent nucleation and simultaneous growth should occur. The nucleation should be fast enough for initially formed nickel clusters to pack tightly onto the substrate surface, and each of these initial clusters should then grow sufficiently to promote three-dimensional (3D) growth. If the growth rate is fast and nucleation is slow, nickel deposits in a more columnar fashion with large, fast-growing crystals and open spaces between the crystals. Under such conditions, highly strained isolated crystals can form, or even large dendrites which can grow through the cathode bags and cause short-circuits (Winand, 1994; Budevski et al., 2000; Kittelty, 2002).
Crucial parameters that need to be controlled in order to optimize the relationship between nucleation and growth during nickel electrodeposition have been established: nickel, sodium sulphate, and boric acid concentrations, temperature, and pH of the sulphate electrolyte (Holm and O'Keefe, 2000; Di Bari, 2010; Crundwell et al., 2011). At sufficiently high nickel concentrations (60 g/L or higher), the conductivity of the electrolyte and mobility of the nickel cations toward the cathode increase. Rapid nucleation with simultaneous growth is promoted and desired uniform, finegrained deposits form preferentially (Abyaneh and Hashemi-Pour, 1994; Ji and Cooper, 1995; Wu et al., 2003). The sodium sulphate concentration is also kept relatively high (between 80 g/L and 100 g/L) to increase the conductivity of the electrolyte. If sodium sulphate is added in large excess, however, the viscosity of the electrolyte increases significantly, which in turn decreases nickel cation activity and mobility (Abyaneh and Hashemi-Pour, 1994; Holm and O'Keefe, 2000; Wu et al., 2003). At higher temperatures (60°C or higher), mobility and conductivity increase, reactions rates are faster, and nickel deposition is promoted. Rounder, low-strained, uniform deposits are formed and current efficiency increases (Lantelme and Seghiouer, 1998; Holm and O'Keefe, 2000; Lupi et al., 2006). The pH of the electrolyte is important for a number of reasons. At low pH (normally controlled between 2 and 3), the formation of nickel hydroxide species (which occurs freely at around pH 5) is limited and pure deposits are expected (Amblard et al., 1983). The pH also plays an important role in controlling the hydrogen reduction reaction (Equation [4]). Boric acid is added as a buffering agent, and it is known that at boric acid concentrations of at least 4 g/L and at low pH (2-3) of the electrolyte, the nickel deposition reaction is promoted and the hydrogen evolution reaction is limited (Armyanov and Sotirova-Chakarove, 1992; Gadad and Harris, 1998). Under these conditions, good quality nickel deposits with low hydrogen pitting, low strain, and good morphology are expected. The mechanism and function of boric acid is still largely unknown and polarization techniques may be able to shed some light on this much-debated subject (Amblard et al., 1983; Tripathy and Das, 2001).
Measurement techniques
Accurate measurement of nucleation and growth in order to evaluate the effect of changes in the crucial parameters is still a challenge. One of the earliest techniques that proved useful in obtaining such information during the electrodeposition process was cyclic voltammetry. This is a potentiodynamic (controlled-potential) polarization technique that measures plating potential (Ep), i.e., the potential at which growth is initiated. This potential is identified by a sudden increase in the cathodic current, as illustrated in Figure 1. The overpotential is sensitive to changes in electolyte parameters, conditions, concentrations, and additives or impurities that may be present in the solution (Kerby et al., 1977; Mackinnon and Brannen, 1977; Warren, 1985; Adcock et al., 2002). This technique, however, requires an intricate and laborious set-up to produce repeatable results and it only provides information regarding changes in the plating potential (Adcock et al., 2002).
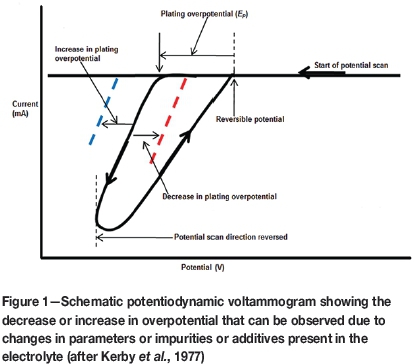
Kerby et al. (1977) developed a successful technique known as the dual-channel continuous electrolyte quality monitor (CEQM), in which overpotential measurements were made at specific current densities for both nucleation and plating by using two individual wires that were monitored simultaneously throughout the electrodeposition process. This technique was laborious and complex, but indicated that nucleation and plating overpotentials could be measured simultaneously. It also established the importance of controlled-current (galvanodynamic) methods compared with classical controlled-potential (potentiodynamic) methods. It was this idea that led to the development, by Adcock et al., (2002), of a new galvanodynamic (current-controlled) measurement technique specifically for zinc electrowinning from sulphate electrolytes. This technique makes use of a three-step galvanodynamic scanning procedure with a very slow initial forward scan rate to measure nucleation overpotential. An intermediate scan rate is then applied after the nucleation point is reached, and a fast scan rate is used on the reverse scan. The plating overpotential is measured at an arbitrary current density (typical of commercial processes). The advantage of using galvanodynamic instead of potentio-dynamic methods is that more detail regarding the initial nucleation is observed and can therefore be measured (Adcock et al., 2002). This technique is illustrated and compared with conventional potentiodynamic voltammetry in Figure 2.
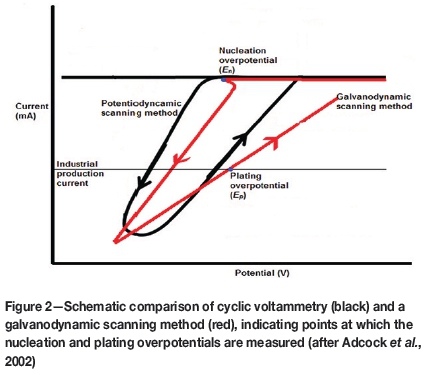
The aim of the current work was to utilize the knowledge gained from the development of this galvanodynamic measurement method (Adcock et al., 2002) to develop a similar method specifically for nickel electrowinning from sulphate electrolyte. The idea was to produce a simple yet reliable measurement tool with the capability of measuring both plating and nucleation potentials accurately and repeatably. This measurement method can then be used to investigate the effect of crucial operating parameters and species concentrations on the electrodeposition process, and ultimately to use the information gained to optimize, monitor, and control commercial nickel electrodeposition operations.
Experimental methods
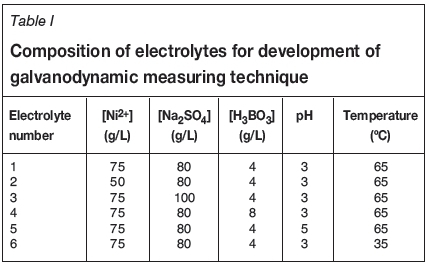
A solution containing 75 g/L nickel, 80 g/L sodium sulphate, and 4 g/L boric acid with a pH of 3 and a temperature of 65°C was used as the standard electrolyte, representative of conditions established that produce acceptable quality nickel deposits. Thereafter, different electrolytes were prepared and tested by changing each of the parameters individually according to Table I.
Electrochemical measurements were performed in a standard three-electrode cell with a Ag/AgCl reference electrode (Metrohm). The cathode consisted of insulated titanium metal (commercial grade) with a surface area of 2.25 cm2. The cathode was connected to conducting wire by silver epoxy glue and cold-mounted in epoxy resin, leaving only the top surface exposed. The surface was then prepared just prior to use by hand-polishing using silicon carbide (SiC) paper from P800 to P1200 while rinsing with deionized water, followed by ultrasonic cleaning in deionized water for 2 minutes. The auxiliary electrode was Pt wire. The auxiliary electrode (anode) and working electrode (cathode) were separated by a polyethylene membrane (product code D21A05, obtained from Ecotao). Fresh electrolyte solution (300 mL) was used for each experiment. The electrolyte was heated throughout each experiment by an encased water jacket with water flowing from a water bath. The temperature of the electrolyte was measured, monitored, and adjusted within ±1°C of the desired temperature using a standard thermometer. The system was connected to a potentio-/galvanostat (Solartron Schlumberger 1286 interface) and a computer with the appropriate software (Corrware and Corrview, obtained from Solartron Analytical).
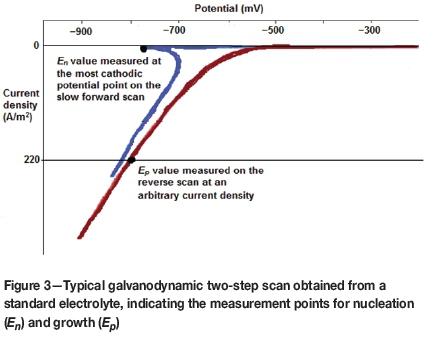
A preferred galvanodynamic scanning method was developed in order to measure nucleation and plating overpotentials accurately and precisely. The newly prepared working electrode was introduced into the cell with prepared electrolyte for 1 minute to stabilize at zero current in galvanostatic mode. Thereafter, two sequential galvano-dynamic scans were applied. The first forward cathodic scan was done at a very slow scan rate (0.1125 mA/s) in order to elucidate the precise turning point at which nucleation started. This was taken as the most cathodic potential point reached during this slow scan. This scan rate was applied from 0 mA to -0.3 mA. The second galvanodynamic scan was then applied from -0.3 mA to -90 mA in the cathodic direction, and then from -90 mA to 0 mA in the anodic direction. This was done to obtain a value for plating overpotential (Ep). It was found that Epwas not as sensitive as nucleation overpotential (E„), and a faster scan rate of 0.5625 mA/s could therefore be used during this second scan. Each scan consisted of a series of small steps in current with the potential measured and recorded at the end of each step. A total of 2000 points was collected and recorded for each two-step process. The nucleation potential (E„) was measured as the most cathodic potential reached on the slow forward cathodic scan. The plating potential (Ep) was measured on the return anodic scan at an arbitrary current density of 220 A/m2, which is commonly used as the applied current density for industrial nickel electrowinning. The scans were repeated in triplicate for each electrolyte. A typical scan indicating the nucleation and growth points is shown in Figure 3.
After each two-step galvanodynamic scan, the program was immediately set to galvanostatic deposition, during which nickel was galvanostatically deposited for 80 minutes at a current density of 220 A/m2 under the same conditions. This was done in order to obtain a thick deposit for morphological, quality, and characteristic evaluation. Electrodeposits were stripped from the substrate surface immediately thereafter, ultrasonically cleaned in deionized water, and mounted for morphological investigation. Each mounted deposit was polished to 1 μΐη diamond finish and then etched for 4 seconds with 50:50 by volume nitric acid:acetic acid. The deposits were then optically inspected and classified according to quality, character, and general morphology. The current efficiency was calculated from the mass of each deposit compared with the theoretical mass determined from Faraday's Law.
Results and discussion
Table II shows the average measurement results from the triplicate scans for each electrolyte specified in Table I. An indication of the accuracy is also given, as well as the current efficiency and a description of the observed quality and morphological characteristics of the produced nickel deposit.
Typical microstructures of cross-sections of thick nickel deposits obtained from the specified electrolytes are shown in Figure 4.
The results show that both nucleation and plating overpotentials can be obtained accurately and repeatably for sulphate electrolytes with varying conditions and parameters by using the developed galvanodynamic technique. The measured overpotential values (both nucleation and plating) are clearly sensitive to and influenced by the conditions and species concentrations in the electrolyte.
If the standard electrolyte (electrolyte 1) is used as a reference point for measured overpotential values and optimum nickel electrodeposition conditions and parameters, a number of observations can be made regarding the other electrolytes. For the electrolyte with low nickel concentration (electrolyte 2), the Enchanged markedly (more negative) and the Eponly slightly (more negative). This suggests that the low concentration of nickel cations is inadequate to promote simultaneous nucleation and growth in order to form closely packed 3D structures. The deposits obtained under these conditions were strained and pitted, which could also indicate promotion of the hydrogen reaction at low nickel concentrations. The current efficiency was much lower compared with standard conditions, suggesting poor plating conditions. A strain crack can be seen in Figure 4b.
At high sodium sulphate concentration (electrolyte 3), both Enand Epvary; 3D growth was promoted and round, uniform, fine-grained, good quality deposits formed (Figure 4c). The relationship between nucleation and growth seemed to be optimum under these conditions. The current efficiency even increased compared with that calculated for electrolyte 1. Much the same observations can be made for deposits obtained from electrolyte 4, which had increased boric acid concentration. The relationship between nucleation and growth once again seemed optimum, even though the Enand Epshifted to more negative values. The exact mechanism of the action of boric acid in the electrolyte is still unclear, but it is apparent that the addition thereof promotes nickel electrodeposition in a desired fashion. No hydrogen pitting was observed in the presence of higher concentrations of boric acid (Figure 4d.
An increase in pH (electrolyte 5) seemed to increase both Enand Ep(became less negative) and the relationship between these two parameters was not ideal. Growth seemed to be promoted without frequent enough nucleation, causing strain, dendrite formation, and severe pitting (Figure 4e). The current efficiency also decreased significantly under these conditions. Similar observations were made for electrolyte 6 (low temperature): very large shifts in Enand Epwere observed while the deposits obtained are strained, cracked, and extremely pitted. The current efficiency was very low.
It is clear that the measured Enand Epvalues as well as the relationship between Enand Epfor each type of electrolyte play a crucial role in the type of growth, quality, and morphological characteristics of the nickel electrodeposit. Future work will further investigate and attempt to classify and correlate the relationship between polarization parameters and morphological outcomes. The developed galvanodynamic technique can also be used to investigate the effects of other variables in the electrolyte, such as additives to promote grain refinement or impurities.
Conclusions
A galvanodynamic polarization measurement technique developed specifically for nickel electrodeposition from typical sulphate electrolytes is presented. This method can be used to measure both Enand Epaccurately, precisely, and repeatably for the various electrolytes tested. The method and experimental set-up are relatively simple and easy to operate. The measured Enand Epvalues are sensitive to changes in electrolyte concentration (nickel or sodium sulphate), the addition of boric acid, and parameters such as pH and temperature. It appears that changes in Enand Ep, as well as the relationship between Enand Ep, vary in a specific way, depending on conditions and parameters of the electrolyte. Therefore, the information gained from measuring the Enand Epcan be useful to anticipate the effect that changes in an electrolyte might have. The technique can be useful in monitoring and control of the nickel electrodeposition process. Under specific conditions, the nickel deposits nucleate and grow in a specific way. It seems desirable to further investigate the relationship between Enand Epvalues measured by this galvanodynamic technique and the morphological and quality characteristics of the nickel deposits to determine any possible correlation.
References
Aaboudi, O., Amblard, J., Chopart, J.-P., and Olivier, A. 2001. A temperature and electrochemical impedance spectroscopy analysis of nickel electrocrystallization from a Watts solution. Journal of Physical Chemistry B, vol. 105. pp. 7205-7210. [ Links ]
Abyaneh, M.Y. and Hashemi-Pour, M. 1994. The effect of the concentration of boric acid on the kinetics of electrocrystallization of nickel. Transactions of the Institute of Metal Finishing, vol. 72, no. 1. pp. 23-26. [ Links ]
Adcock, P.A., Adeloju, S.B., and Newman, O.M.G. 2002. Measurement of polarization parameters impacting on electrodeposit morphology I: Theory and development of technique. Journal of Applied Electrochemistry, vol. 32. pp. 1101-1107. [ Links ]
Amblard, J., Froment, M., Maurin, G., Spurellis, N., and Trevisan-Souteyrand, E. 1983. Nickel electrocrystallization - from nucleation to textures. Electrochimica Acta, vol. 28, no. 7. pp. 909-915. [ Links ]
Armayanov, S. and Sotirova-Chakarove, G. 1992. Hydrogen desorption and internal stress in nickel coatings obtained by periodic electrodeposition. Journal of the Electrochemical Society, vol. 139, no. 12. pp. 3454-3457. [ Links ]
Budevski, E., Staikov, G., and Lorenz, W.J. 2000. Electrocrystallization nucleation and growth phenomena. Electrochimica Acta, vol. 45. pp. 2599-2574. [ Links ]
Crundwell, F., Moats, M., Ramachandran, V., Robinson, T., and Davenport, W. 2011. Extractive Metallurgy of Nickel, Cobalt and Platinum-Group Metals. Elsevier. pp. 327-346. [ Links ]
Di Bari, G.A. 2010. Electrodeposition of nickel. Modern Electroplating, 5th edn. Schlesinger, M. and Paunovic, M. (eds.). Wiley, Hoboken, New Jersey. pp. 79-114. [ Links ]
Gadad, S. and Harris, T.M. 1998. Oxygen incorporation during the electrodeposition of Ni, Fe and Ni-Fe alloys. Journal of the Electrochemical Society, vol. 145, no. 11. pp. 3699-3703. [ Links ]
Holm, M. and O'Keefe, T.J. 2000. Evaluation of nickel deposition by electrochemical impedance spectroscopy. Journal of Applied Electrochemistry, vol. 30. pp. 1125-1132. [ Links ]
Ji, J. and Cooper, W.C. 1995. Surface pH measurements during nickel electrode- position. Journal of Applied Electrochemistry, vol. 25. pp. 642-650. [ Links ]
Kerby, R., Jackson, H., O'Keefe, T., and Wang, Y. 1977. Evaluation of organic additives for use in zinc electrowinning. Metallurgical Transactions B, vol. 8B. pp. 661-612. [ Links ]
Kittelty, D. 2002. The electrocrystallization of nickel and its relationship to the physical properties of the metal. PhD thesis, Murdoch University, Australia. [ Links ]
Lantelme, F. and Seghiouer, A. 1998. Model of nickel electrodeposition from acidic medium. Journal of Applied Electrochemsitry, vol. 28, no. 9. pp. 907-913. [ Links ]
Lupi, C.M., Pasquali, M., and Dell'Era, A. 2006. Studies concerning nickel electrowinning from acidic and alkaline electrolytes. Minerals Engineering, vol. 19. pp. 1246-1250. [ Links ]
Mackinnon, D.J. and Brannen, J.M. 1977. Zinc deposit structures obtained from high purity synthetic and industrial acid sulfate electrolytes with and without antimony and glue additions. Journal of Applied Electrochemistry, vol. 7. pp. 451-459. [ Links ]
Munoz, A.G., Salinas, J.B., and Bessone, J.B. 2003. First stages of Ni deposition onto vitreous carbon from sulfate solutions. Thin Solid Films, vol. 429. pp. 119-128. [ Links ]
Tripathy, B.C. and Das, S.C. 2001. Effect of Mg2+, Li+, Na+ and K+ pt on the electrocrystallization of nickel from aqueous sulfate solutions containing boric acid. Journal of Applied Electrochemistry, vol. 31, no. 5. pp. 573-577. [ Links ]
Warren, I.H. 1985. The application of polarization measurements in the control of zinc tankhouse operation. Proceedings of Zinc '85. Mining and Metallurgical Institute of Japan. pp. 251-264. [ Links ]
Winand, R. 1994. Electrodeposition of metals and alloys - new results and perspectives. Electrochimica Acta, vol. 39, no. 8/9. pp. 1091-1105. [ Links ]
Wu, R., Oliazadeh, M., and Alfantazi, A.M. 2003. Electrical conductivity and density of NiSO4/H2SO4 solutions in the range of modern nickel electrore- fining and electrowinning electrolytes. Journal of Applied Electrochemistry, vol. 33. pp. 1043-1047. [ Links ]
This paper was first presented at the 3rd Young Professionals Conference, 9-10 March 2017, Innovation Hub, Pretoria, South Africa.


{kind=link}