










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkJournal of the Southern African Institute of Mining and Metallurgy
On-line version ISSN 2411-9717
Print version ISSN 2225-6253
J. S. Afr. Inst. Min. Metall. vol.116 n.6 Johannesburg Jun. 2016
http://dx.doi.org/10.17159/2411-9717/2016/v116n6a4
PAPERS - COOPER COBALT AFRICA CONFERENCE
Study of the dissolution of chalcopyrite in solutions of different ammonium salts
T. Moyo; J. Petersen
Mineral to Metal Research Initiative, University of Cape Town, Cape Town, South Africa
SYNOPSIS
The oxidative leaching of chalcopyrite in ammoniacal solutions has been evaluated using electro-analytical techniques and controlled bulk leaching studies. The anodic dissolution process has been established to be a seven-electron transfer process under nitrogen in ammonia-ammonium sulphate solutions and ammonia-ammonium carbonate solutions, which suggests that the sulphur is oxidized to thiosulphate and the copper and iron released as Cu+ and Fe2+ in these systems. The deportment of Fe2+ and S2O32- is affected by choice of the ammonium salt used in the leaching process. In the perchlorate salt, only five electrons are transferred, supporting formation of different sulphur species as proposed for the sulphate and carbonate salts. Scanning electron microscopy and energy-dispersive spectroscopic analysis of the mineral surface after leaching indicate presence of an iron-sulphur surface layer completely free of copper in the sulphate system, an iron-rich surface layer in the perchlorate solutions, and absence of surface layer build-up in carbonate solutions. XRD analysis of a bulk leach residue from leaching in ammonia-ammonium sulphate solutions showed the surface layer to be mostly amorphous (90%). The crystalline content (10%) is composed of 95% polymorphs of anhydrous iron oxide hydroxide FeO(OH). Choice of ammonium salt and the hydrodynamic environment of leaching have been shown to influence the presence or absence of the surface product, as well as its nature.
Keywords: chalcopyrite, ammonia, surface deposits, coulometry, iron oxy-hydroxide
Introduction
Hydrometallurgical processes for copper extraction provide a viable route to recover the metal from mixed and low-grade ores, as well as overcome the environmental challenges faced by the traditional pyrometallurgical processes. Chalcopyrite is not only the most abundant of the copper sulphides, but also the most stable, making it recalcitrant to hydrometallurgical processes. Hence, hydrometallurgical processing of chalcopyrite continues to be an attractive area of research due to the vaguely understood surface chemistry of the mineral in different aqueous media. Different routes for hydrometallurgical treatment of chalcopyrite can be followed. These include thermal treatment prior to leaching, direct leaching and direct electrochemical leaching (Venkatachalam, 1991). Direct leaching of chalcopyrite can be carried out in various solution systems, as reviewed by Roman and Benner (1973) and Venkatachalam (1991). Ammoniacal solutions are attractive and effective lixiviants that form stable amine complexes with some base metal cations while rejecting iron. Leaching of chalcopyrite in ammoniacal solutions in the presence of an oxidant is possible due to the stabilization of copper (I) and copper(II) by ammonia at elevated pH levels. In oxygenated ammonia solutions, it has been suggested that chalcopyrite dissolves according to Equation [1] (Beckstead and Miller., 1977a):
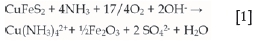
Chalcopyrite is characterized by very slow leaching kinetics and this has been strongly linked to the formation of a passive film on its surface. Researchers have not reached a consensus on the actual composition or degree of stability of this passive surface film. Fe2O3 or its hydrated form Fe2O3.xH2O (Beckstead and Miller, 1977b; Yin et al., 1995; Forward and Mackiw, 1955; Feng and Van Deventer, 2002) and Fe(OH)3 (Yin et al., 1995; Warren and Wadsworth, 1984) have been reported to be the surface products of chalcopyrite oxidation. Warren and Wadsworth (1984) studied the electrochemical oxidation of chalcopyrite in ammonia-ammonium sulphate solutions and reported significant amounts of ferrous iron (3-20%) in the product film. The authors postulated that a ferrous iron intermediate was formed, which can be readily oxidized to ferric, and the oxidation can be achieved even if only traces of dissolved oxygen are present. If ferrous iron is formed from chalcopyrite dissolution, it is expected that its further reactions would be affected by the solution conditions, hence the different surface effects as observed under the carbonate and sulphate ammonium salts. Asselin (2011) reported that Fe(II) ammines are thermodynamically stable only under reducing conditions and concluded that they were unlikely to be formed if oxygen is present. He presented quasi-equilibrium Pourbaix diagrams for the Fe-NH3-H2O system. According to the diagram, Fe(OH)3 is the species present at noble potentials across all pH ranges, while Fe(OH)2 is present only above pH 11.
Forward and Mackiw (1955) suggested that sulphide minerals, chalcopyrite in this case, react with oxygen, water, and ammonia to produce soluble salts in such a way that the iron present in each mineral particle is converted to hydrated iron oxide in situ, with the result that the particles, when leaching is complete, consist of hydrated iron oxide pseudo-morphic with the original mineral. Beckstead and Miller (1977b) discuss the nucleation and growth of the haematite passivating layer (in ammonia-ammonium sulphate solutions), stating that this occurred on the anodic sites, thus limiting the overall reaction by limiting the anodic reaction. Agitation at speeds up to 3000 r/min increased the rate of reaction, possibly by abrading the haematite layer and exposing more of the anodic sites. The contradiction between these two studies stems from the fact that Beckstead and Miller looked at the nucleation and growth of the Fe product and identified it as haematite, while Forward and Mackiw looked at 'an ion substitution mechanism, resulting in a Fe product atypical of chalcopyrite'. In a study on the surface oxidation of chalcopyrite in alkaline solutions, Yin et al. (2000) reported that the iron in the top layer of chalcopyrite oxidized, forming a monolayer of Fe(OH)3 and Fe2O3, while the copper and sulphur remained unoxidized in the original chalcopyrite crystal structure, forming a phase they designated as CuS2*, which together with the Fe(OH)3 and Fe2O3 retarded the oxidation of the mineral. The abrasion of the passivating layer, although not widely researched in ammoniacal media, has been demonstrated to have potential to increase copper recoveries and a process, the FLSmidth® Rapid Oxidative Leach (ROL), has been patented (Eyzaguirre et al., 2015). This process uses very low-energy interstage attrition to enhance dissolution of copper-bearing minerals, primarily chalcopyrite, through a mechano-chemical process that is based on the removal of the passivating product layers. The application of such a process agrees well with results from Beckstead and Miller's (1977b), in which the chalcopyrite surface was observed to be free of surface deposit at high agitation speeds, possibly due to increased abrasion.
Literature on iron chemistry in ammonia-ammonium carbonate solutions is limited for the chalcopyrite leaching process, thus reference will be made to the Caron process. Studies have been carried out on the leaching of nickel from pre-reduced laterite ores (nickel and iron are present as reduced metallic grains and metal alloys) (Das and Anand, 1995; Jandová and Pedlík, 1994, Kim et al., 1991; Lee, Osseo-Asare, and Pickering, 1985; Nikoloski, 2002; Nicol, Nikoloski, and Fittock, 2004). At a pH of about 9.8 in ammoniacal solutions, the dominant dissolved iron species is the ferrous tetra-ammine ion and the dissolution reaction has been postulated to occur according to Equation [2] (Nikoloski, 2002; D'Aloya and Nikoloski, 2012; Subrata, 2010; Nikoloski and Nicol, 2006; Osseo-Assare and Asihene, 1979):
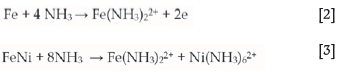
The ferrous ammine is oxidized in aerated solutions to form ferric, which does not form ammines and is thus precipitated as Fe(OH)3 releasing the ammonia:
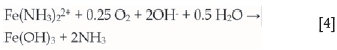
Nikoloski (2002) showed that iron is prone to passivation in solutions typical of those used in practice, and confirmed its occurrence in leaching reactors of a commercial scale in the Caron process. Passivation was shown to occur due to the formation of an oxide layer at the potentials that can be attained in the presence of high concentrations of dissolved oxygen.
An earlier study by the authors (Moyo et al., 2015) investigated chalcopyrite dissolution in ammonia-ammonium sulphate solutions. Coulometric measurements on a chalcopyrite electrode resulted in postulating a seven-electron transfer mechanism, with the potential formation of thiosulphate as the product of sulphur oxidation. Copper(II), rather than oxygen, was found to be the primary oxidant on the mineral surface. Rapid formation of an iron oxide layer on the electrode surface was observed, which did not inhibit, but slowed down, the rate of anodic dissolution over time. No iron was found in solution, whereas no copper was found in the surface layer.
The present study aims to elucidate the nature of the surface deposits and to what extent anions in the ammoniacal solution might have an effect on their formation. Tests were done in ammonium sulphate, carbonate, and perchlorate solutions. Coulometric tests were done in each case to ascertain if the dissolution mechanism remained the same in the different solutions before investigating the surface layers formed on the chalcopyrite electrode.
Experimental
Chalcopyrite electrodes
Samples of natural chalcopyrite from Durango, Mexico (supplied by Wards Natural Science) were mounted on brass stubs with conductive silver epoxy and the assembly then imbedded in non-conductive epoxy resin. X-ray diffraction (XRD) analysis of the mineral sample indicated it to be 100% chalcopyrite, while quantitative electron microscopy (QEMSCAN™) indicated it to be 95% chalcopyrite, with 3% sphalerite and 1% calcite. The electrode surface was polished on 1200-grit SiC abrasive paper, then on 1 μΐη, 0.3 μΐη, and 0.05 μηι aluminium oxide prior to each experiment, except where stated. Only one electrode was used for the purposes of this study and the exposed surface area of the electrode was measured to be 0.36 cm2 using imagej™ for image analysis. The surface area was measured intermittently during the course of the study and was found not to change significantly.
Electrochemical measurements
A standard three-electrode cell with a chalcopyrite rotating-disk working electrode was used for all electrochemical measurements. A saturated calomel reference electrode and platinum wire auxiliary electrode were used. A freshly polished chalcopyrite electrode was placed into an electrolyte of desired composition and rotated at 1600 r/min. Rest potentials were measured for 30 minutes. A Gamry Series G 300/750 potentiostat was used. Coulometric experiments were carried out by fixing the potential at a value measured during the rest potential measurements described above (after 30 minutes) in a solution of similar composition but in the absence of copper(II) ions. Total charge was obtained by integration of the current-time curves. The current response of the electrode was then measured for 2 hours. Additional coulometric experiments were carried out for 5 hours or 22 hours at the rest potential or the vicinity of the rest potential in a minimum volume of solution. At the end of the experiment, the solutions were analysed for copper using inductively coupled plasma optical emission spectroscopy (ICP-OES). All potentials are reported against the standard hydrogen electrode (SHE) unless stated otherwise. Experiments were conducted at 25°C in a thermostatted cell containing 30 mL solution. The total Cu concentration in solution at the end of the coulometric experiments was established by ICP-OES.
Controlled leaching experiments
Three tests were carried out, two in which blocks of chalcopyrite 5 mm x 5 mm x 5 mm were leached (reactors A and B) and a third one in which micronized chalcopyrite (81% passing -75 μm) was leached in 3 M total ammonia-ammonium sulphate solutions at 1% solids (reactor C). Reactor A had glass beads added, which allowed for the abrasion of the surface product from the chalcopyrite blocks, while reactor B had no beads in it. The leaching experiments went on for a period of 5 days, allowing for significant amounts of surface product to be generated. Magnetic stirrers were used; this was adequate to keep the sample in swirling motion but could not suspend the sample in solution for reactors A and B, while the micronized sample in reactor C was effectively suspended. The lixiviant was first placed in the reactors and brought to the desired reaction temperature, 25°C. The temperature was controlled by a thermostatted water bath circulating water around the jacketed reactors. Oxygen was bubbled into the solution for 10 minutes prior to the experiment, allowing the solution to be saturated with dissolved oxygen, and a blanket of oxygen gas was maintained over the solution through the entire experiment. Solution pH was 9.6±0.15 from the buffer of the ammonia-ammonium sulphate. The mineral sample was then placed into the reactor, marking the onset of the reaction. Solution samples were taken intermittently and analysed for copper using atomic absorption spectrophotometry (AAS).
Solution preparation
Deionized water and reagent-grade CuSO4.5H2O, NH4OH, (NH4)2SO4, (NH4)2CO3, NH4ClO4, H2SO4, and NaOH were used. All solutions were prepared by mixing 1:1 molar ratio ammonium hydroxide to ammonium salt based on the ammonium ion, i.e., a 1 M (NH3+NH4+) solution was made by mixing 1 M NH4OH with 0.5 M (NH4)2SO4. Rest potentials that were later used for the coulometric experiments, were measured in the presence of 5 g/L initial copper(II). Either oxygen (99.99%) or nitrogen was bubbled into the electrolyte for 10 minutes prior to starting the experiment, and bubbling was continued throughout the experiment, taking care to ensure that gas bubbles did not accumulate on the chalcopyrite electrode surface (in electrochemical experiments). This was established to be sufficient time to either saturate the solution with dissolved oxygen or remove it from solution.
Surface measurements
The surface of the chalcopyrite was examined by scanning electron microscopy (SEM) and QEMSCAN. An elemental analysis was done on the surface using energy-dispersive X-ray spectroscopy (EDS) in an attempt to characterize the surface effects and products under the conditions of this study. The leach residue was further analysed using X-ray diffraction (XRD), Brunauer-Emmett-Teller (BET) surface analysis, as well as digested and subjected to chemical analysis using atomic absorption spectroscopy.
Results and discussion
Coulometry
Coulometry studies were done in the different salt media, as described in the methodology. The stoichiometry of the anodic dissolution reaction was determined by this method. The coulometric results shown in Table I include data from our previous study (Moyo et al., 2015) on the sulphate system. Table I shows the number of electrons transferred per molecule of chalcopyrite (measured as copper released into solution) at potentials corresponding to rest potentials measured in 1 M (NH3+NH4+) with 5 g/L Cu(II) at 25'C, but here in the absence of initial Cu in solution. All measurements were repeated at least three times and the calculated number of electrons transferred was found not to vary by more than 5%.
In the carbonate system, as previously found for the sulphate system, approximately seven electrons are transferred per mole of copper. This suggests the formation of a thiosulphate intermediate and that the copper and iron are in the cuprous and ferrous states. The cuprous and ferrous ions would then be subsequently oxidized in solution, in non-faradaic reactions, to the cupric and ferric state. The stoichiometry of the reaction would be approximated by Equation [5]. In perchlorate solution, on the other hand, approximately five electrons are transferred, suggesting the potential formation of elemental sulphur and the formation of cupric and ferric (Equation [6]), i.e., the oxidation of the iron and the copper take place as faradaic reactions:
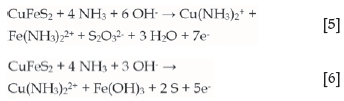
Topological effects
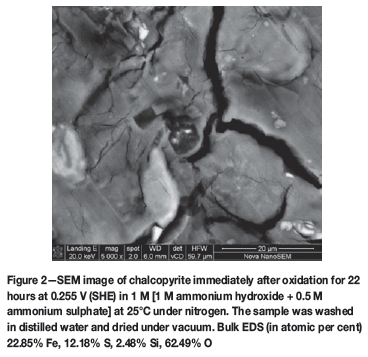
The visual differences between the chalcopyrite surfaces after polarization were apparent even to the naked eye, with those treated in the sulphate and perchlorate solutions having lost their lustre and exhibiting a reddish-brown colouration, while that treated in the carbonate solution had lost its lustre but did not exhibit any colour changes. Figure 1A shows optical microscope images of a freshly polished chalcopyrite surface and 1B shows chalcopyrite after oxidation in ammonia-ammonium sulphate solutions - the difference is quite clear. Freshly polished chalcopyrite prior to leaching was analysed using SEM (image not shown). Atomic percentages, 24.62% Cu, 25.67% Fe and 49.71% S, measured by bulk EDS, are consistent with those of pure chalcopyrite. This provides a basis of comparison for the samples analysed post-leaching. Figure 2 shows a SEM image of a chalcopyrite surface after 22 hours of oxidation in ammonia-ammonium sulphate solutions under nitrogen. It is copper-free and has significant amounts of sulphur and iron. EDS shows the surface composition to be 22.85 at.% Fe, 12.18 at.% S, 2.48 at.% Si, and 62.49 at.% O. The Fe:S ratio is 1.9:1, which represents a significant shift from the 1:2 ratio of pure chalcopyrite. The presence of sulphur on the surface was previously reported by Warren and Wadsworth (1984), who upon scraping off and dissolving the surface film from their experiments identified some orthorhombic sulphur, confirming that elemental sulphur was present in both the surface film and leachate. The researchers attributed this to the dispropor-tionation of some of the intermediate sulphur species expected to be found in the leachate. Kuhn, Arbiter, and Kling (1974) presented the possibility of the recrystallization of sulphur from the sulphide to form rhombic or monoclinic sulphur, which would be expected to be found in localized sites within the mineral, while cations diffuse out of the mineral. Therefore, the presence of sulphur on the mineral surface, as observed in this study, is possible although not widely reported.

The morphology of the surface layer suggests that it may have been formed by precipitation, which is in contradiction to the ion-substitution mechanism proposed by Forward and Mackiw (1955). This is more apparent when looking at residue on the surface of a chalcopyrite block leached for 5 days in the absence of glass beads (reactor B) (Figure 5), which shows what appears to be an agglomerate of small particles. This surface product readily dissolved upon short contact with concentrated sulphuric acid solutions, leaving behind an etched but pure chalcopyrite surface. The acid solution used to wash off the surface layer was analysed using ICP and found to contain 16.05 mg/L iron but only 0.095 mg/L copper, confirming the SEM results which indicated it to be an iron-rich surface layer.
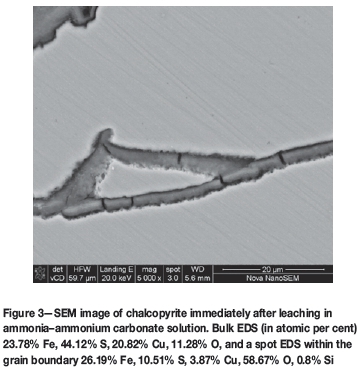


As stated in the introduction, the presence of a relatively small amount of sulphur in the surface deposit prompted the investigation of chalcopyrite dissolution in non-sulphate media. Figure 3 shows the mineral surface after 22 hours of oxidation in ammonia-ammonium carbonate solution. All other solution conditions were maintained the same. The bulk EDS results indicates 23.78% Fe, 44.12% S, 20.82% Cu, and 11.28% O. Copper accounts for 20.82% of the surface. Assuming that this comes from the chalcopyrite then, in terms of atomic ratios, this agrees well with the amount of sulphur present, leaving a small proportion of excess iron, i.e., 2.96 percentage points more iron than copper. The ratio of Fe:S is 1:1.85, fairly close to that of pure chalcopyrite. This suggests that only a very thin film of an iron-rich phase would be present. The visible cracks were observed to be more copper-depleted. EDS analysis of the crack areas showed only 4-6% to be copper, suggesting these to be areas of preferential chalcopyrite dissolution in the carbonate medium. This is further supported by the visibility of polishing marks on the unleached areas in Figure 3. The differences in surface effects between the sulphate and carbonate solutions suggest that the surface reaction mechanism varies, although the same number of electrons is transferred. Iron carbonate complexes potentially form in this system, and tend to stay in solution, whereas the formation of a surface precipitate in the sulphate system is more likely to be linked to the formation of iron sulphate complexes that precipitate.
The behaviour of iron in ammonium carbonate solutions has been explored by some researchers, with the focus on improving the leaching kinetics in the Caron process. Kim et al. (1991) studied the active-passive behaviour of sintered iron in ammoniacal ammonium carbonate solutions at pH 9.7 and reported that surface films were formed on the bulk iron during exposure to air or immersion in ammoniacal solutions. The authors characterized the surface films using X-ray photoelectron spectroscopy (XPS) and generated cathodic reactivation transients; they reported that in the presence of air Fe3O4 was formed. Caldeira, Ciminelli, and Osseo-Asare (2008) investigated the effect of carbonate on pyrite oxidation in alkane solutions and identified (using diffuse reflectance infrared spectroscopy) iron carbonate compounds as one of the products of pyrite oxidation. The authors explain that the increased oxidation rate typically observed on pyrite in carbonate solutions is possibly due to the formation of Fe(II)-CO3 complexes, the buffering effect of the carbonate and the fact that complexation with bicarbonate/carbonate provides a stronger Fe(III)/Fe(II) redox couple, increasing the Fe(III) solubility. This could be an explanation for the apparent lack of significant surface layer formation on chalcopyrite in the carbonate system, through the possible formation of soluble Fe(II)-CO3 complexes which are then transported away from the vicinity of the mineral surface. It should be noted that, although other studies of the carbonate system reported the presence of an oxide layer, the obvious lack of any significant layer in this study could also be attributed to the hydrodynamics around the electrode area. Rotating at 1600 r/min may be sufficient to allow the transport of the metastable iron carbonates away from the mineral surface before they have a chance to nucleate and precipitate. The effect of electrode rotation speed on the extent of surface layer build-up and its morphology in the sulphate system have been reported by Beckstead and Miller (1977b), who found that chalcopyrite surfaces were much cleaner and relatively free of surface product at agitation speeds higher than 1100 r/min. They postulated that increased agitation speeds improved leaching by increasing turbulence, which in turn allowed for the abrasion of the surface product, thereby exposing a fresh mineral surface for leaching.
To provide further assessment, especially with regard to the sulphur deportment, tests were done in ammonia-ammonium perchlorate solutions, as the perchlorate ions are not expected to complex with the oxidation products of chalcopyrite.
Figure 4 shows an SEM image of a chalcopyrite sample after 22 hours of leaching in ammonia- ammonium perchlorate solutions. Visually, the surface appears not dissimilar to that observed in the sulphate system. However, the bulk surface EDS gave an Fe:S atomic ratio of 1:1.1 and copper is present at a S:Cu of 1:0.38. Analysis of isolated flaky areas on the sample showed these areas to be more iron-rich: Fe:S is 2:1 whereas the S:Cu remains at 1:0.38. The results suggest the presence of an iron-rich surface together with a sulphur species. The atomic ratios of the sulphur and iron are not consistent with the stoichiometric ratios suggested by coulometry (Equation [6]), which would require all sulphur to accumulate on the surface as elemental sulphur and hence there should be twice as much sulphur as iron. However, tests done so far cannot be used to make any conclusive deductions as to whether the sulphur observed to be present on the mineral surface is elemental sulphur or some form of Fe-S complex. Furthermore, the fact that copper was retained on the surface could have affected the copper balance during the coulometric study of the perchlorate system, thus leading to an under-estimate of electrons transferred.
Experiments were done in which 5 mm blocks of the same sample of chalcopyrite were leached in ammonium sulphate, as described in the methodology. In both instances, the remaining pieces of the blocks, as well as any residue that accumulated in the reactor at the end of the experiment, were washed in distilled water, dried, and analysed under the microscope and by QEMSCAN. Figure 5 shows a SEM image of the residue collected from the reactor that contained the beads. Neither sulphur nor copper were found on the residue, and EDS of the residue gave a ratio of O:Fe 1.1:1, fairly close to the 1.5:1 ratio typical of haematite, although this is unlikely to form under the present experimental conditions.
XRD analysis of the residue indicated it to be 90% amorphous, and of the 10% crystalline content 95% comprised polymorphs of anhydrous iron oxide hydroxide FeO(OH) as shown in Table II. ICP and LECO analysis indicated the residue to contain 44% iron and less than 1% of both copper and sulphur.
It should be noted that the leach reaction proceeded much faster in the reactor containing glass beads, with copper recoveries at the end of the 5-day leaching period at 54.9%, whereas in the reactor without glass beads merely 14.7% was leached. It is apparent that the continuous removal of the surface deposits in the reactor with beads had a positive impact on the recoveries. This is in agreement with Beckstead and Miller (1977b), who reported that at low stirring speeds the rate of the reaction was significantly reduced: they related this to the growth and nucleation of a haematite phase. They also reported on the possible impact of stirring speed on the morphology of the haematite phase they observed. In the development of the Sherritt-Gordon process, critical agitation speeds were reported (Forward and Mackiw, 1955) and these are said to have allowed for the abrading of the 'haematite' layer, thus improving leaching rates. Also, in the Arbiter process (Kuhn et al., 1974), intense mixing is used to abrade the haematite phase from the Cu-Fe-S phase, thus exposing a fresh surface for reaction, as well as achieving good oxygen transfer rates.
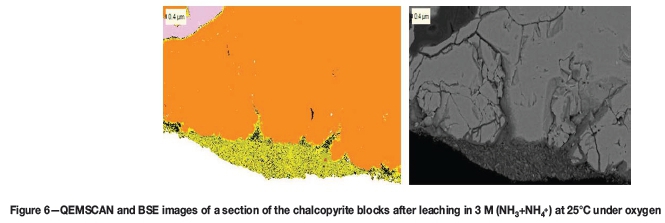
Figure 6 shows QEMSCAN and SEM images of one of the sample blocks placed in the reactor without beads. An iron-rich surface deposit is visible at the edge on the mineral, and the morphology of this surface layer can be seen on the BSE image to be similar to that of the debris in Figure 5. The bulk phase, orange in colour, has been identified as chalcopyrite, while the yellow is an iron-rich phase containing small percentages of sulphur and the black phase is an iron- and oxygen-rich phase without any sulphur. An EDS analysis of the surface layer showed it to be 57.7% Fe, 32.12% O, 1.45% S, and the rest reported to be silica and calcium.
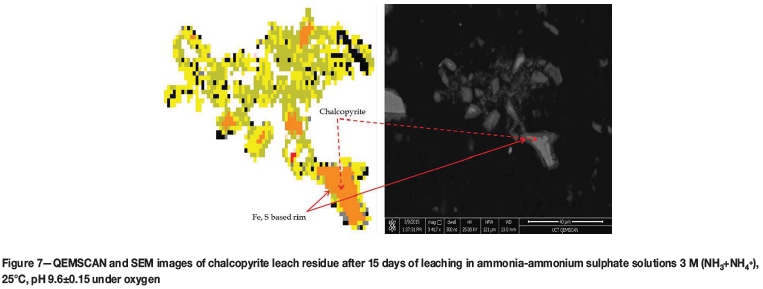
Leach residue from reactor C (finely ground material) was washed, dried, and analysed. Figure 7 shows a QEMSCAN and SEM image of part of the debris. It is apparent that an iron-sulphur-based layer formed around a chalcopyrite core. The layer also contained iron which is not associated with sulphur, which was identified as an iron oxy-hydroxide species. It is worth mentioning that all species that contained iron and sulphur in Fe:S ratios higher than 0.5 reported to pyrite and pyrrhotite, but this classification does not in any way mean this phase is in actual fact pyrite or pyrrhotite.
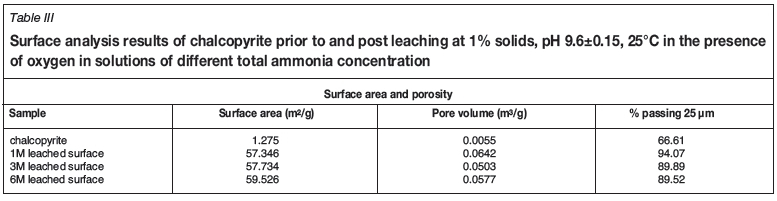
From Figures 5 to 7, the morphology of the surface product appears to be significantly different from that of the initial sample. The layer appears to be composed of an agglomeration of smaller particles as to it be relatively porous compared to chalcopyrite. Surface area and porosity were measured using the Brunauer-Emmett-Teller (BET) method on the micronized sample prior to and after leaching (Table III). It is apparent that the surface area and surface pore volume of leached particles increased significantly relative to that of the non-leached chalcopyrite. It is also notable that the percentage of particles less than 25 μηι (diameter) increased significantly in the leach residue samples, from 67% in an unleached sample to about 90% in leached samples. This suggests that although oxidation of chalcopyrite may occur in situ as suggested by Forward and Mackiw (1955), this surface layer is in fact quite friable and would be easily abraded, hence the formation of smaller sized particles, also confirmed by the increased surface area per gram of the samples. These observations are consistent with findings by Guan and Han (1997), who identified a friable and porous surface product while oxidizing chalcopyrite in ammonia-ammonium iodide solutions. The readily soluble nature of the surface layer observed when washing the electrode layer in acid solutions indicates that this surface layer is not stable.
Conclusions
1. The number of electrons (seven) transferred per mole copper during anodic oxidation is similar for the ammonia-ammonium sulphate and ammonia-ammonium carbonate solutions
2. Ammonia-ammonium perchlorate solutions promote a five-electron transfer/copper reaction, possibly forming elemental sulphur on the mineral surface
3. Ammonium sulphate leaching results in the formation of a Fe-oxyhydroxide layer with low sulphur on the mineral surface
4. Ammonia-ammonium carbonate solutions resulted in marginal accumulation of iron on the mineral surface, but no formation of a layer was observed
5. Ammonium perchlorate leaching results in the formation of a Fe-oxyhydroxide layer with moderate sulphur on the mineral surface
6. The surface product was largely amorphous (90%) and significantly more porous (9-12 times) than unleached chalcopyrite. The observed morphology of the surface product suggests that it is formed through secondary precipitation rather than as part of the chalcopyrite dissolution mechanism
7. Surface abrasion allows for the removal of the surface product, leading to improved leaching recoveries
8. The abraded surface product from the small particles leaching experiment contained no sulphur, while surface products found on the stationary block of mineral contained small quantities of sulphur.
It has thus been shown that a relatively unstable iron product forms on the surface of chalcopyrite through secondary reactions to the faradaic oxidation reaction. This product may contain small percentages of sulphur but, regardless of system, the majority of leached sulphur reports to the solution. Choice of ammonium salt and the hydrodynamic environment of leaching influence the presence or absence, as well as the nature, of the surface product. It appears that the formation of surface products in turn influences the reaction mechanism of chalcopyrite dissolution, and the two aspects need to be studied in conjunction.
Acknowledgements
The authors would like to acknowledge the Centre for Bioprocess Engineering Research, the Centre for Imaging and Analysis at the University of Cape Town, and Professor M.J. Nicol for their contribution to this work.
This project has been funded through the Minerals to Metals Research Initiative, which is supported by the South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation (NRF) of South Africa. Any opinion, finding, and conclusion or recommendation expressed in this material is that of the authors and the NRF does not accept any liability in this regard.
References
Asselin, E. 2011. Thermochemistry of the Fe, Ni and Co-NH3-H2O system as they relate to the Caron process: a review. Minerals and Metallurgical Processing, vol. 28, no. 4. pp. 169-175. [ Links ]
Beckstead, L. and Miller, J. 1977a. Ammonia, oxidation leaching of chalcopyrite-reaction kinetics. Metallurgical and Materials Transactions B, vol. 8, no. 1. pp. 19-29. [ Links ]
Beckstead, L. and Miller, J. 1977b. Ammonia, oxidation leaching of chalcopyrite-surface deposit effects. Metallurgical and Materials Transactions B, vol. 8, no. 1. pp. 31-38. [ Links ]
Caldeira, C.L., Ciminelli, V.S.T., and Osseo-Asare, K. 2008. Pyrite oxidation in alkaline solutions: the carbonate effect. Hydrometallurgy 2008: Proceedings of the Sixth International Symposium.. Young, C.A., Taylor, P.R., Anderson, C.G., and Choi, Y. (eds.). Society for Mining, Metallurgy and Exploration, Littleton, CO, USA. pp. 990-999. [ Links ]
D'Aloya, A. and Nikoloski, A.N. 2012. The passivation of iron in ammoniacal solutions containing copper (II) ions. Hydrometallurgy, vol. 111-112. pp. 58-64. [ Links ]
Das, R.P. and Anand, S. 1995. Precipitation of iron oxides from ammonia-ammonium sulphate solutions. Hydrometallurgy, vol. 38, no. 2. pp. 161-173. [ Links ]
Eyzaguirre, C., Rocks, S., Klepper, R., Baczek, F., and Chaico, D. 2015. The FLSmidth® Rapid Oxidative Leach (ROL) Process: A mechano-chemical approach for rapid metal sulphide dissolution. Graber, T., Taboada, M.E., and Valenzuela, F. (eds). Proceedings of the 7th International Seminar on Process Hydrometallurgy, Hydroprocess 2015, 22-24 July 2015. Gecamin, Santiago, Chile. [ Links ]
Feng, D. and Van Deventer, J.S.J. 2002. Leaching behaviour of sulphides in ammoniacal thiosulphate systems. Hydrometallurgy, vol. 63, no. 2. pp. 189-200. [ Links ]
Forward, F.A. and Mackiw, V.N. 1955. Chemistry of ammonia pressure process for leaching Ni, Cu, and Co from Sherritt Gordon sulphide concentrates. Transactions AIME, Journal of Metals. pp. 457-463. [ Links ]
Guan, Y. and Han, K. 1997. The leaching kinetics of chalcopyrite (CuFeS2) in ammonium iodide solutions with iodine. Metallurgical and Materials Transactions B, vol. 28, no. 6. pp. 979-985. [ Links ]
JandovA, J. and PedlIk, M. 1994. Leaching behaviour of iron-nickel alloys in ammoniacal solution. Hydrometallurgy, vol. 35, no. 1. pp. 123-128. [ Links ]
Kim, H.S., Kho, Y.T., Osseo-Asare, K., and Pickering, H.W. 1991. Active and passive behaviour of sintered iron in ammoniacal ammonium carbonate solution. Metallurgical Transactions B, vol. 22, no. 3. pp. 323-332. [ Links ]
Kuhn, M.C., Arbiter, N., and Kling, H. 1974. Anaconda's Arbiter process for copper. CIM Bulletin. pp. 6762-6773. [ Links ]
Lee, J.W., Osseo-Asare, K., and Pickering, H.W. 1985. Anodic dissolution of iron in ammoniacal ammonium carbonate solution. Journal of the Electrochemical Society, vol. 132, no. 3. pp. 550-555. [ Links ]
Moyo, T., Petersen, J., Franzidis, J.-P., and Nicol, M.J. 2015. An electrochemical study of the dissolution of chalcopyrite in ammonia-ammonium sulphate solutions. Canadian Metallurgical Quarterly, vol. 3. pp. 368-27. [ Links ]
Nicol, M.J., Nikoloski, A.N., and Fittock, J.E. 2004. A fundamental study of the leaching reactions involved in the Caron process. Proceedings of the International Laterite Symposium. Imrie, W.P. and Lane, D.M. (eds.). The Minerals, Metals and Materials Society, Warrendale, PA. pp. 369-384. [ Links ]
Nikoloski, A.N. 2002. The electrochemistry of the leaching of pre-reduced nickel laterites in ammonia-ammonium carbonate solution. PhD thesis, Murdoch University, Perth, Australia. [ Links ]
Nikoloski, A.N. and Nicol, M.J. 2006. The electrochemistry of the leaching reactions in the Caron process 1. Anodic processes. Electrochemical Society Transactions, vol. 2, no. 3. pp. 197-207. [ Links ]
Osseo-Asare, K. and Asihene, S.W. 1979. Heterogeneous equilibria in ammonia/laterite leaching systems. Proceedings of the International Laterite Symposium. Evans, D.J.I., Shoemaker, R.S., and Vetman, H. (eds.). Society of Mining Engineers of the American Institute of Mining, Metallurgical, and Petroleum Engineers, New York. pp. 585-609. [ Links ]
Subrata, R. 2010. Electrochemical dissolution and passivation behaviour of iron in ammoniacal Caron leaching solution. MSc thesis, University of British Columbia, Canada. [ Links ]
Warren, G. and Wadsworth, M. 1984. The electrochemical oxidation of chalcopyrite in ammoniacal solutions. Metallurgical and Materials Transactions B, vol. 15, no. 2. pp. 289-297. [ Links ]
Yin, Q., Kelsall, G.H., Vaughan, D.J., and England, K.E.R. 1995. Atmospheric and electrochemical oxidation of the surface of chalcopyrite (CuFeS2). Geochimica et Cosmochimica Acta, vol. 59, no. 6. pp. 1091-1100. [ Links ]
Yin, Q., Vaughan, D.J., England, K.E.R., Kelsall, G.H., and Brandon, N.P. 2000. Surface oxidation of chalcopyrite (CuFeS2) in alkaline solutions. Journal of the Electrochemical Society, vol. 147, no. 8. pp. 2945-2951. [ Links ] ♦
This paper was first presented at the, Copper Cobalt Africa Conference, 6-8 July 2015, Avani Victoria Falls Hotel, Victoria Falls, Livingstone, Zambia.


{kind=link}
{kind=link}
{kind=link}
{kind=link}