










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkJournal of the Southern African Institute of Mining and Metallurgy
On-line version ISSN 2411-9717
Print version ISSN 2225-6253
J. S. Afr. Inst. Min. Metall. vol.116 n.6 Johannesburg Jun. 2016
http://dx.doi.org/10.17159/2411-9717/2016/v116n6a3
PAPERS - COOPER COBALT AFRICA CONFERENCE
Xanthate chemisorption at copper and chalcopyrite surfaces
J.L. Bowden; C.A. Young
Metallurgical and Materials Engineering, Montana Tech, Butte MT, USA
SYNOPSIS
Cyclic voltammetry experiments were conducted on copper and chalcopyrite (CuFeS2) in the absence and presence of ethyl xanthate, and compared to mass-balanced EH-pH diagrams. The results for copper duplicate those found in the literature and confirm xanthate chemisorption. However, the, results indicate that chalcopyrite oxidizes to chalcocite (Cu2S) and only afterwards is chemisorption observed due to small currents appearing with xanthate. This phenomenon suggests that the mineral's hydrophobicity is induced by more than dixanthogen and copper xanthate. Hydrophobicity was found to be pH-dependent over a range of alkaline conditions (pH 7-11) at narrow potentials (0 to -200 mV). An EH-pH diagram for chalcopyrite with xanthate is presented to illustrate the conditions under which chalcopyrite would be hydrophobic and thereby more thoroughly explain the results in the literature.
Keywords: copper, chalcopyrite, xanthate, chemisorption, isotherms, voltammetry
Introduction
Sulphide minerals are generally concentrated from ores by froth flotation, a physiochemical process that induces separation based on differences in hydrophobicity. In this process, a gas is bubbled into a slurry and the minerals that are hydrophobic (water-repelling) adhere to the bubbles and float to the surface while the hydrophilic (water-loving) minerals remain in the slurry. With sulphide minerals, the hydrophobicity is generally established in two ways. The first is by the addition of a collector, which is a heteropolar molecule with a reactive inorganic head group and an inert organic tail. Typically, the head group bonds with the sulphide mineral surface by adsorbtion either chemically (chemisorption) or physically (physisorption) or by complexing with atoms that solubilize somewhat at the surface (surface precipitation). These interactions leave the organic tail protruding from the mineral surface, thereby inducing hydropho-bicity. The second method is by oxidation of the mineral surface to form a layer that is metal-deficient/sulphide-rich or comprised of elemental sulphur. Because the layer mimics naturally hydrophobic sulphur, this enables 'collectorless' flotation. Furthermore, this oxidation phenomenon adds an electrochemical dimension to the understanding of sulphide mineral flotation.
In this regard, the literature on sulphide mineral flotation abounds with studies conducted in the absence and presence of collector, typically involving electrochemical experiments such as cyclic voltammetry, galvanostatic polarization, and impedance measurements coupled with thermodynamic speciation calculations typically plotted as EH-pH/Pourbaix diagrams. Most studies involve xanthate as collector but have included, for example, mercaptans, mercaptobenzothiazoles, and dithiophosphates. Many were also accompanied by analytical measurements involving various spectroscopic tools, including (but not limited to) Fourier transform infrared spectroscopy (FTIR), Raman spectroscopy, X-ray photoelectron spectroscopy/electron spectroscopy for chemical analysis (XPS/ESCA), atomic force microscopy (AFM), mineral liberation analysis/quantitative scanning electron microscopy (MLA/QEM-SEM), and time-of-flight secondary-ion mass spectrometry (ToF-SIMS) (Smart et al., 2014). Although these studies have allowed the conditions under which sulphide minerals float to become better understood, there still remains uncertainty as to how the collectors interact with the mineral surfaces. 'Surface electrochemistry' knowledge allows flotation separations of sulphide minerals to be made more efficiently and with improved selectivity.
Typical copper sulphide minerals include covellite (CuS), chalcocite (Cu2S), chalcopyrite (CuFeS2), bornite (Cu5FeS4), enargite (Cu3AsS4), tennantite (Cu12AsS13), and tetrahedrite (Cu12SbS13). Of these, chalcopyrite is the most common. Most studies involving chalcopyrite flotation were conducted with and without xanthate. Results show distinctions between a chemisorbed layer and a secondary co-adsorbed layer of xanthate components corresponding to surface-precipitated copper xanthate and physisorbed dixanthogen (Fuerstenau, 2007). These species are respectively illustrated by the following reactions:

where X represents the xanthate molecule. However, it has been debated whether there exists a region of chemisorbed xanthate (Suoninen and Laajalehto, 1993):

and whether the region results in an increased flotation recovery of chalcopyrite (Suoninen and Laajalehto, 1993). It is important to point out that the oxidation reactions leading to dixanthogen (Equation [2]) and chemisorbed xanthate (Equation [3]) only increase the complexity of the surface electrochemistry involved.
The purpose of this investigation was to study the surface electrochemistry of chalcopyrite with xanthate at various EH potentials and pH values in the hope of observing xanthate chemisorption. To do this effectively, it was felt that experiments with a known system should also be conducted and, in regard, copper was first investigated.
Experimental
Cyclic voltammetry
Cyclic voltammetry (CV) experiments were conducted with an EG&G Princeton Applied Research 263A potentiostat/ galvanostat. A standard three-electrode system was employed with the reference electrode being Ag/AgCl and the counter-electrode being graphite. Working electrodes were made from either electrical grade copper wire obtained from VWR Scientific or natural chalcopyrite from Butte, MT, provided by the mineral museum at Montana Tech. MLA/SEM revealed that the copper wire was pure and the chalcopyrite contained minor amounts of quartz inclusions but not more than 5%. All potentials are reported as SHE/EH potentials. Typical scans in the absence of xanthate were initiated at 0 mV and scanned negatively to a lower potential of -1000 mV and then reversed positively to +1000 mV for as many as six cycles. Copper voltammograms in both the absence and presence of xanthate were initiated between -240 and -440 mV in increments of 25 mV, scanned negatively to a lower potential of -600 mV, and then positively to the final potential of -240 mV. Chalcopyrite voltammograms in the absence and presence of xanthate were restricted to -50 mV as the initial and upper potentials and -600 mV as the lower potential. For all experiments, the initial potential was held constant for approximately 5 minutes and scan rates of 15 mV/s were used. In addition, 1 L electrochemical cells were used with 500 mL solutions and 5-minute nitrogen purges. Solutions were buffered at pH values between 7 and 12.
Materials
Working electrodes were mounted in polyvinylchloride (PVC) tubing using epoxy and polished between CV scans using 400- grit paper. To fit in the tubing, the copper wire was wound into a cylindrical shape and chalcopyrite was cut into approximate dimensions of 3x3x8 mm with the 3x3-mm face being the electrode surface. Electrodag was used to connect the chalcopyrite to the wiring needed to bridge the circuit to the potentiostat. The pH buffers were made up from reagent-grade chemicals of potassium and sodium phosphate as well as sodium carbonate and bicarbonate, all from Fischer Scientific. Potassium ethyl xanthate (KEX) was formulated using potassium hydroxide, ethanol, and carbon disulphide obtained from VWR Scientific following the procedure of Nedjar et al. (2009).
EH-pH diagrams
EH-pH diagrams were generated with StabCal (Gow et al., 2015; Huang, 2015) using thermodynamic data from Forssberg et al. (1988) for the copper/xanthate/water and chalcopyrite/xanthate/water systems.
Results and discussion
Copper
Cyclic voltammetry
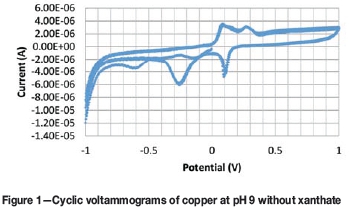
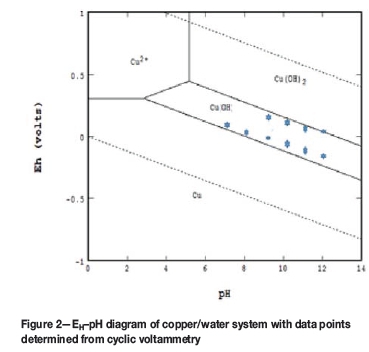
A typical CV scan for copper is shown in Figure 1. In this example, results were obtained at pH 9 and illustrate that copper oxidizes at approximately 0.0 V and again at 0.2 V, as indicated by the increase in anodic current as the scan progresses from negative to positive voltage. When these values are plotted on the EH-pH diagram of the copper/water system in Figure 2, it is clear that 0 V corresponds to Cu oxidation to CuOH (equivalent to Cu2O) and that 0.2 V correlates with CuOH oxidation to Cu(OH)2 (equivalent to CuO). The dashed lines in the diagram illustrate the conditions where water is stable. When this is repeated for the CVs determined at the other pH values, the results also compare quite well with the stability lines between the species on the EH-pH diagram. Because of the good correlation between the thermodynamically predicted and experimentally obtained results, it is concluded that the wire is an effective copper electrode.
Chemisorption
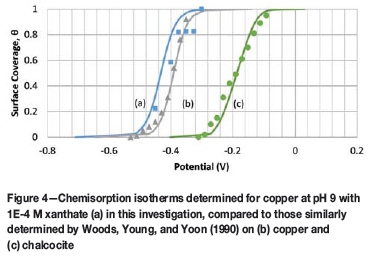
Woods et al. (1990) developed a technique to calculate adsorption isotherms for chemisorbed xanthate on copper and chalcocite (Cu2S). With this technique, they obtained CVs in the absence and presence of xanthate by holding the initial potential constant for 5 minutes and then collecting the CV. Afterwards, the initial potential was incrementally changed until the complete range at which chemisorption occurred was covered and no change was observed between the tests performed with and without xanthate. By integrating the area covered by each CV and dividing by the scan rate, the charge passed for each CV was calculated. Subtracting the charge without xanthate from the charge with xanthate at each potential yielded the charge caused solely by xanthate chemisorption. When all charges were divided by the maximum charge obtained, chemisorption isotherms resulted with fractional surface coverage, Θ, plotted as a function of potential.
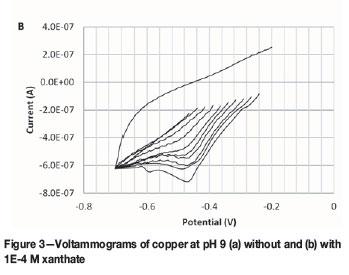
To validate the work of Woods et al. (1990) and simultaneously establish a procedure for testing on chalcopyrite, we repeated their technique for copper. Voltammograms are presented in Figure 3a without xanthate and Figure 3b with xanthate. In Figure 3, the voltammograms determined with an initial potential near -0.5 V are the same, thereby explaining why no xanthate chemisorption was calculated at this potential (see Figure 4). Likewise, the voltammogram determined with an initial potential of -0.24 V in Figure 3b shows a second cathodic peak appearing near -0.60 V, which is caused by the surface precipitation of copper xanthate (see Equation [1]). In this regard, it is concluded that xanthate chemisorption is at a maximum at -0.265 V and therefore yields complete monolayer coverage ( i.e., θ = 1) at this potential. Likewise, the other voltammograms in Figure 3 yielded fractional coverages between these values (i.e., 0 < θ < 1).

As with Woods et al. (1990), it is assumed the resulting chemisorption isotherm follows the Frumkin equation in the form adopted by Schultze (1980):
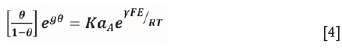
where θ is the fractional surface coverage, aA is the activity of the adsorbate, g and K are constants, and y is the electrosorption valency. Assuming the activity is equal to the xanthate concentration, [X ], the following equation is determined:
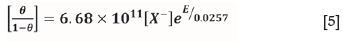
which agrees well with the chemisorption isotherm determined by Woods et al. (1990):
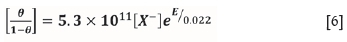
Chalcopyrite
Cyclic voltammetry
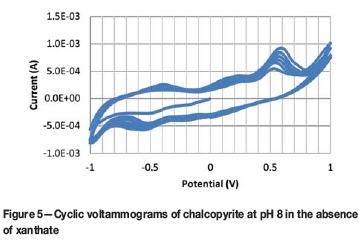
An example CV for chalcopyrite in the absence of xanthate is shown in Figure 5. In this case, results were obtained at pH 8 and the anodic portions show that chalcopyrite oxidizes at approximately -0.75 V, -0.05 V, and 0.35 V. When these values are plotted on the EH-pH diagram of the chalcocite/water system in Figure 6, it is clear that the potentials correspond to oxidation of Cu to CuFeS2 to Cu2S to CuOH to Cu(OH)2, respectively. Oxidation is also observed at 0.8 V but was not plotted because this correlated with oxygen evolution and therefore is not relevant. Although there is more overall scatter in this data, it does match reasonably well with the thermodynamic calculations. Of course, it is understood that chalcopyrite is a semiconductor (unlike copper being a pure conductor) and electrochemical reactions are partially irreversible as a result.
Because chalcocite is a product of chalcopyrite oxidation, it is reasonable to expect that xanthate chemisorption on chalcopyrite would occur in regions similar to copper and chalcocite (see Figure 4). Furthermore, because chalcopyrite/xanthate flotation is predominantly accomplished near pH 9, initial testing was done at pH 9 such that the starting potential was changed from 0 to -0.3 V in increments of 0.05 V until the reaction was no longer seen. Xanthate chemisorption was no longer observed at potentials below -0.25 V, the same potential shown in Figure 4 and observed by Young et al. (1991) for chalcocite (Cu2S) in a separate study. However, an isotherm could not be ascertained because the results appeared to depend on the prior amount of chalcocite that formed. Similar tests were repeated at pH 7 and 11 but in these cases no chemisorption was observed.
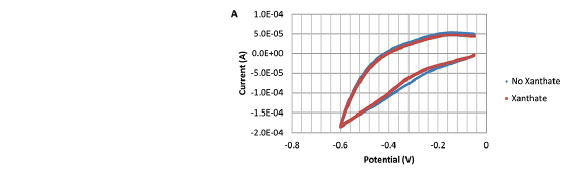
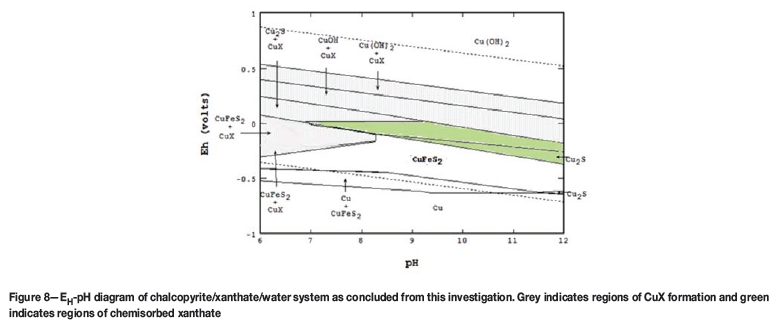
Rather than incrementally change the initial potential, tests were repeated at a single initial potential of -0.05 V. A lower potential of -600 mV was used to ensure the charge due to xanthate chemisorption could be determined. Results in the absence and presence of xanthate for the three pH values of 7, 9, and 11 are shown in Figure 7a-c, respectively. Results for pH 7 and 11 clearly show that the CVs with and without xanthate are the same, illustrating that xanthate chemisorption is indeed absent. However, at pH 9, there appears to be significant current flow in both the absence and presence of xanthate. According to the EH-pH diagram in Figure 8, at -0.05 V, CuFeS2 is stable at pH 7 (no current was observed), Cu2S is stable at pH 9 (current was observed), and CuOH is stable at pH 11 (no current was observed). Clearly, xanthate chemisorption does not occur on CuFeS2 and CuOH but does occur on Cu2S, even if it is an oxidation product of chalcopyrite. This suggests that chalcopyrite flotation can be attributed to dixanthogen and copper xanthate surface precipitate as well as chemisorbed xanthate. Unfortunately, these thermodynamic calculations do not show dixanthogen being co-stable with chalcopyrite. In fact, dixanthogen predominates only at potentials more oxidizing than the stability region of CuX.


Chemisorption
The collectorless flotation of chalcopyrite can be attributed to elemental sulphur formation and is depicted by the following reaction, which also yields chalcocite:
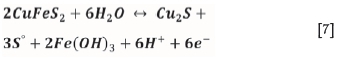
In order to estimate the charge passed by this reaction, the CVs without xanthate in Figures 7a and 7b were integrated and divided by the scan rate. Because the charge passed in Figure 7a did not change with xanthate, it is assumed that it emanates from IR drop and is the same as that passed in Figure 7b without xanthate. The difference between the two therefore yields the charge passed due solely to chalcocite formation. Based on the molar charge of electrons being 9.65x104 coulombs and the reaction producing six moles of electrons, an estimated 1.95x10-9 moles of chalcocite form. Likewise, the difference in charges determined for the two CVs in Figure 7b yields the charge passed due solely to xanthate chemisorption on the resulting chalcocite. In this regard, an estimated 7.98x10-10 moles of chemisorbed xanthate formed. According to Liu (2014), the area of surface that is accessible for intermolecular contact in the S-S atoms of a xanthate molecule is 19.2 Å2. In a Cu2S lattice, there is approximately one Cu atom per 9 Å2 according to the studies by Evans (1979). From this data, the surface area of the Cu2S available for adsorption was found to be 2.11x10-4 m2 and the surface area of xanthate formed was found to be 9.23x10-5 m2. By dividing the amount of chemisorbed xanthate by the amount of Cu2S formed, a surface coverage of over 40% is calculated. Since Woods et al. (1990) noted that 20% coverages yielded 50% chalcocite recoveries and 50% coverages yielded 90% chalcocite recoveries, a 40% coverage would be adequate to yield a high flotation recovery.
Luttrell and Yoon (1984) showed that chalcopyrite will start to float (defined as 10% recovery) at pH 10 near -100 mV vs SHE in the absence of collector. Woods (1996) obtained similar results near 0 mV vs SHE at pH 11. By comparison, when Roos et al. (1990) added 1.5x10-4 M KEX, they found that the onset of chalcopyrite flotation occurred near -150 mV vs SHE at pH 9.2. Similarly, Chander (2003) floated chalcopyrite ore with various collectors and found flotation did not occur until potentials exceeded 0 mV vs SHE at pH 10.5-11. These EH-pH data points plot near the shaded region in Figure 8, with two points close to the border at low potentials, commonly referred to as the lower flotation edge. However, it is strongly suggested that Equation [7] indicates elemental sulphur will likely be present as well, making it difficult to differentiate between collectorless flotation and xanthate flotation caused by chemisorption on chalcocite (see Equation [3]). Furthermore, even though Equation [3] is pH-independent, xanthate cannot chemisorb unless the surface it forms at is present. Hence, the pH-dependency is due directly to chalcocite being produced as a result of Equation [7]. This would help explain why the data from these studies is scattered (Luttrell and Yoon, 1984; Woods, 1996; Roos et al., 1990; Chander, 2003).
In an attempt to more definitively prove that a layer of chemisorbed xanthate was forming on the surface of the chalcopyrite, in situ and ex situ test work with Raman spectroscopy was performed. Spectra were generated for mineral samples at pH 9 and 0 V vs SHE both with and without xanthate in solution. The results did not definitively show the chemisorbed xanthate species. Work in the Metallurgical and Materials Engineering Department at Montana Tech is continuing along these lines.
Conclusions
Although it is known that xanthate can be used to increase the flotation recovery of chalcopyrite there is still uncertainty as regards the exact nature of the surface chemistry and all the areas that xanthate might be interacting with the chalcopyrite surface. This research shows evidence that the chemisorption of xanthate onto a chalcopyrite surface will occur in regions where Cu2S formed upon oxidation of the chalcopyrite surface. At pH 9 it was shown that this reaction occurs between -0.05 and -0.25 V versus SHE, similar to what Woods et al. (1990) observed on chalcocite. To help validate the study, previous experiments of Woods et al. (1990) on copper were also validated. The results are corroborated by flotation results noted in the literature.
Acknowledgements
The authors thank Newmont Metallurgical Services and Newmont Mining Corporation for their generous support for this research. Newmont's goal of advancing mineral processing and extractive metallurgical knowledge by investing in graduate research at Montana Tech is greatly appreciated.
References
Chander, S. 2003. A brief review of pulp potentials in sulfide flotation. International Journal of Mineral Processing, vol. 72, no. 2. pp. 141-150. [ Links ]
Evans, H.T. 1979. The crystal structures of low chalcocite and djurleite. Zeitschriftfur Kristallographie, vol. 150. pp. 299-320. [ Links ]
Forssberg, K.S.E., Antti, B-M., and Palsson, B.I. 1984. Computer-assisted calculations of thermodynamic equilibria in the chalcopyrite-ethyl xanthate system. Reagents in the Minerals Industry. Jones, M.J. and Oblatt, R. (eds). Institute of Mining and Metallurgy, London, UK. pp. 251-264. [ Links ]
Gow, R., Huang, H., and Young, C. 2015. Utility of mass balanced Eh-pH diagrams II: stoichiometry of the Cu-As-S-H2o system. Mineral and Metallurgical Processing Journal [in press]. [ Links ]
Huang, H.H. 2015. STABCAL, Version 9.1., Montana Tech, Butte, MT, USA. [ Links ]
Fuerstenau, M.C., Chander, S., and Woods, R. 2007. Sulfide mineral flotation. Froth Flotation: A Century of Innovation. Fuerstenau, M.C., Jameson, G., and Yoon, R.H. (eds.). Society for Mining, Metallurgy and Exploration, Littleton, CO, USA. pp. 425-465. [ Links ]
Liu, J. Wen, S, Deng, J., Chen, X., and Feng, Q. 2014. DFT study of ethyl xanthate interaction with sphalerite (1 1 0) surface in the absence and presence of copper. Applied Surface Science, vol. 311. pp. 258-263. [ Links ]
Luttrell, G.H. and Yoon, R.H. 1984. Surface studies of the collectorless flotation of chalcopyrite. Colloids and Surfaces, vol. 12. pp. 239-254. [ Links ]
Nedjar, Z., Bouhenguel, M., and Djebaili, A.E. 2009. Synthesis and structural characterization of xanthate (KEX) in sight of their utilization in the processes of sulfides flotation. Journal of Minerals and Materials Characterization and Engineering, vol. 9. pp. 469-477. [ Links ]
Roos, J.R., Celis, J.P., and Sudarsono, A.S. 1990. Electrochemical control of metallic copper and chalcopyrite-xanthate flotation. International Journal of Mineral Processing, vol. 28. pp. 231-245. [ Links ]
Schultze, J.W. 1980. The role of elementary steps in the kinetics of adsorption processes. Proceedings of the Third Symposium on Electrode Processes. Bruckenstein, S., McIntyre, J.D.E., Miller, B., and Yeager, E. (eds.). The Electrochemical Society, Princeton, NJ, USA. pp. 167-189. [ Links ]
Smart, R.S.C., Gerson, A.R., Hart, B.R., Beattie, D.A., and Young, C.A. 2014. Innovation in measurement of mineral structure and surface chemistry in flotation: past, present, and future. Mineral Processing and Extractive Metallurgy: 100 Years of Innovation, Anderson, C.G., Dunne, R.C., and Uhrie, J.L. (eds). Society for Mining, Metallurgy and Exploration, Littleton, CO, USA. pp. 577-602. [ Links ]
Suoninen, E. and Laajalehto, K. 1993. Structure of thiol collector layers on sulphide surfaces; XVIII International Mineral Processing Congress. Australasian Institute of Mining and Metallurgy, Carlton, Victoria, Australia. pp. 625-629. [ Links ]
Woods, R. 1996. Chemisorption of thiols on metal and metal sulphides. International Journal of Mineral Processing, vol. 29. pp. 401-453. [ Links ]
Woods, R., Young, C.A., and Yoon, R.H. 1990. Ethyl xanthate chemisorption isotherms and Eh-pH diagrams for the copper/water/xanthate and chalcocite/water/xanthate systems. International Journal of Mineral Processing, vol. 30. pp. 17--3. [ Links ]
Young, C.A., Basilio, C.I., and Yoon, R.H. 1991. Thermodynamics of chalcocite-xanthate interactions. International Journal of Mineral Processing, vol. 31. pp. 265-279. [ Links ] ♦
This paper was first presented at the, This paper was first presented at the, Copper Cobalt Africa Conference, 6-8 July 2015, Avani Victoria Falls Hotel, Victoria Falls, Livingstone, Zambia.


{kind=link}
{kind=link}