










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkJournal of the Southern African Institute of Mining and Metallurgy
versão On-line ISSN 2411-9717
versão impressa ISSN 2225-6253
J. S. Afr. Inst. Min. Metall. vol.116 no.1 Johannesburg Jan. 2016
http://dx.doi.org/10.17159/2411-9717/2016/v116n1a9
PAPERS OF GENERAL INTEREST
An algorithm for determining kinetic parameters for the dissociation of complex solid fuels
P. Mavhengere; T. Vittee; N.J. Wagner; S. Kauchali
School of Chemical Engineering, University of Witwatersrand, Johannesburg, South Africa
SYNOPSIS
An established distributed activation energy model (DAEM)-based algorithm for the dissociation of complex fuels obeying linear kinetics was modified to determine the kinetic parameters of materials reacting in a CO2 gas stream by incorporating the random pore reaction model (RPM). The algorithm was adapted to the RPM and was able to derive the activation energy, E, the grouped pre-exponential factor, A, and the number of reactions occurring in the thermal conversion process. Furthermore, the mass fraction associated with each unique reaction was obtained. The ability to accurately determine multiple reactions and changes in the kinetic parameters during the reaction distinguishes the algorithm as a unique and robust method for determining kinetic parameters for the pyrolysis of complex fuels. The novelty in this research was the adaptation of the RPM and other reaction models to the DAEM algorithm, and hence to other conversion processes. The algorithm was tested on simulated conversion data and experimental data from thermogravimetic analysis of the dissociation of a South African coal char and a 50:50 (wt%) coal-biomass blend char under CO2 atmosphere. The specific mass fraction of the reactive material dissociating under a particular set of kinetic parameters was determined, and all sets of data were successfully modelled to high accuracy.
Keywords: distributed activation energy model, random pore reaction model, kinetics, gasification
Introduction
Fossil fuels are currently the world's primary energy source. Coal is the most abundant low-cost fossil fuel, but the high carbon to hydrogen ratio results in production of large quantities of CO2 which, due to concern over global warming, is an unfavourable consequence of the use of this fuel (Irfan, Usman, and Kusakabe, 2011). Coal gasification is one of the most versatile and cleanest ways to convert coal to fuel and energy (Research and Markets, 2006). The ability of the gasification process to eliminate and/or contain air pollutants and greenhouse gases (GHGs) makes this process highly favourable. Hence, it is vital that this process be thoroughly understood to optimize its advantages over conventional coal conversion processes.
One particular aspect of the gasification process which is considered in this research is the determination of the kinetic parameters of the reactions occurring when biomass and coal chars are gasified. A comprehensive review of the many factors affecting the kinetics of gasification in CO2 is given by Irfan, Usman, and Kusakabe (2011). Various methods of determining kinetic parameters have been proposed and explored over the years, with varying degrees of success. Of particular interest is collection of methods which are commonly known as model-free/integral iso-conversion methods (Starink, 2003). While the model-dependent methods (Rotaru and Goêa, 2009) are based mainly on the Langmuir-Hinshelwood model, the iso-conversion methods are based on the premise that reaction rate is a product of two functions: one depending solely on temperature, and the other depending solely on the fraction transformed. This is demonstrated by Equation [1].

The temperature-dependent function generally assumes Arrhenius dependency (Starink, 2003):

In Equation [2], E is the activation energy (kJ/mol), A is the grouped pre-exponential factor (m-1s-1), R is the universal gas constant (8.314 J/(mol K)), and T is the reaction temperature (K). By substituting and integrating Equations [1] and [2], for non-isothermal conversion at a constant heating rate (β), Equation [3] is obtained.
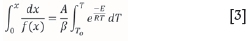
The right-hand side expression of Equation [3] is known as the 'temperature integral'. Integral iso-conversion methods make an approximation to the temperature integral1 and then find E. Since the iso-conversion methods evaluate E for the reaction independent of the reaction model, various methods must further be applied to determine the rest of the kinetic triplet. The kinetic triplet consists of the reaction model function, (f(x)), where x is conversion; E, the activation energy of the conversion reaction; and A, the grouped pre-exponential factor. Iso-conversion methods are usually followed by discrimination procedures for identifying the true conversion function of each linear non-isothermal process (Rotaru and Goêa, 2009; Vyazovkin, 2008). Note that some of these methods are applicable to single step conversions where the E evaluated does not show systematic variation with x (Vyazovkin, 2008). For heterogeneous fuels like coal, where multiple step processes occur, a different approach must be taken. Such scenarios are considered in the distributed activation energy model (DAEM)-based algorithm presented in the current paper.
The presented method evaluates the Emodel independently, as an iso-conversion method. Upon the evaluation of E, a reaction model is assumed, and is used to evaluate the corresponding A. The algorithm provides a way of inverting the DAEM, resulting in the determination of discreet mass-fraction values and accurate identification of E and A for each parallel step participating in the thermal decomposition of the fuel. The current study focuses on extending this method to conversions best described by the random pore reaction model (RPM).
The algorithm developed by Scott et al. (2006a) is based on the DAEM. Coal was first treated as a mixture of a large number of species decomposing by parallel first-order reactions by Pitt (1962). The DAEM makes use of this description, and further assumes that the complexity of the fuel is such that a continuous distribution of Evalues exists for a discretized number of arbitrary reactions. This allows a function of E, and time, to define the mass of volatile material with E values between the initial activation energy (E0) and the activation energy at some point in time (Et) (Scott et al., 2006a). The assumption leads to the formation of a double exponential term, which acts over a narrow range of E values and changes as time progresses (Please, McGuinness, and McElwain, 2003). According to Please, McGuinness, and McElwain, this term is the main source of numerical difficulty in application of the DAEM. Scott et al. (2006a) describe this term as given by Equations [4] and [5].
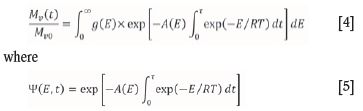
Here, Mv(t) is the total mass of volatile matter, Mvois the initial value of Mv(t), g(E) is the underlying initial distribution of E which characterizes the material, and g(E) may be evaluated from:

Please, McGuinness, and McElwain (2003) outline a number of numerical approximations to the evaluation of Equation [4]. Different approaches to the evaluation of the initial distribution of Ehave been adopted; for example, a Gaussian distribution has been assumed to characterize the distribution of E by some authors (e.g. Kirtania and Bhattacharya, 2012). Numerous approximations have also been suggested to give a closed form of the double integral term. An approximate closed form of the model can be obtained by assuming that, as conversion proceeds, the functional groups with the lowest Evalues (i.e. weakest bonds) dissociate first, as demonstrated by Rostami, Hajalgol, and Wrenn (2004). Other approaches were taken by various authors, as illustrated by Scott et al. (2006a). From these, it is noted that Miura and Maki (1998) showed that it is possible to evaluate the A and E of a particular reaction assumed to dominate at a given conversion by comparing the same volatile yield in two or more experiments performed at different heating rates (Scott et al., 2006a). Braun and Burnham (1987) also demonstrated the first discretization of the distribution of E. This was carried out by assuming a finite set of discrete first-order reactions in the place of the continuous distribution of Eoutlined in Equation [6]. Using these approaches to solving the DAEM, Scott et al. (2006a) proposed an algorithm based on this discretization. This algorithm will be referred to as Scott's algorithm. Scott's algorithm was developed to determine the pyrolysis kinetics of complex solid fuels using the first-order reaction model. Coals and biomasses are complex fuels due to their heterogeneous nature. In the current study, the algorithm was adapted to the RPM; due to the complicated form of the RPM, it was selected for demonstration of the algorithm's capability to be adapted to other reaction models. A general format for adaptation of the model is also outlined in this study. Many authors have proven the DAEM to be a successful method of determining the kinetic parameters of the pyrolysis of complex fuels (e.g. Kirtania and Bhattacharya, 2012; Scott et al., 2006a, 2006b; Miura and Maki, 1998; Maki, Takatsuno, and Miura, 1997). The application of the DAEM to other conversion processes is set to benefit the study of the gasification and combustion processes of complex heterogeneous fuels.
Algorithm development
The original DAEM-based algorithm was derived and shown to be effective in determining the kinetic parameters for pyrolysis (Scott et al., 2006a, 2006b). The algorithm is applied in three main steps, where E, A, and the mass fraction,f; for a candidate reaction, i, are calculated. The original algorithm is presented in Equation [7]:
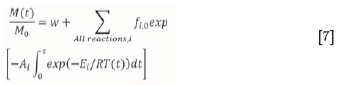
where M(t) is the sample mass of initial value M0containing a fraction w of inert material, and f(i,o) is the initial mass fraction of M0which decomposes with activation energy Ei and pre-exponential factor Ai. The double-exponential term can be expressed as WtNote thatftrepresents the discrete fractional density function, or remaining mass fraction as a function of conversion. The full derivation of the algorithm can be found in Scott et al. (2006a). The double-exponential term in Equation (7) is identified as:
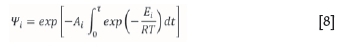
which, if it is multiplied by a constant heating rate, can be expressed as:
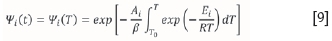
Like the DAEM, the algorithm proposes that the reactions in a complex solid fuel occur in parallel and assumes that at each fractional conversion, a single reaction is dominant. The algorithm uses this assumption and the expression in Equation [9] to calculate E without the use of a proposed reaction model. At this point, the method is akin to an iso-conversion method; significant differences between this method and iso-conversion versions appear later.
The first step of the algorithm considers the conversion at two different heating rates, say β1and β2. If it is then assumed that the ithreaction is the only reaction taking place at this conversion, thenf =f (^2,T2). It then follows, since Ψ is also a function of heating rate, that:

Then, substituting the heating rate and taking natural logarithms on both sides yields:
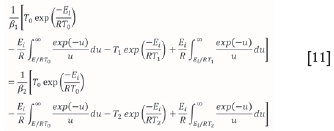
From this, Eican be determined using temperature-conversion data and the heating rates used. This result is presented as a conversion range with corresponding Ei values. Since this step is model-independent, no modification is required.
Once Eihas been found using Equation [11], Aícan be evaluated (Scott et al., 2006a). It is assumed that the reaction of component i is at its maximum rate of decomposition at the particular conversion. This assumption follows from how the approach is posed, i.e that at each fractional conversion, only one reaction is dominating. The assumption is best explained by Miura (1995). Miura demonstrates how the Ψί (T) function changes steeply with E at a given temperature. This then allows the Ψi(T) function to be assumed by a step function (U) at a particular activation energy Essuch that:

This approximation then corresponds to the assumption that only one reaction (with activation energy Es) is occurring at a particular temperature, T (Miura and Maki, 1998; Miura, 1995). The assumption was found to hold at an approximate Esvalue evaluated at Ψi(E, T) = 0.58 for many combinations of the distribution curve and the pre-exponential factor.
Based on this assumption, the value of conversion x that results in this maximum decomposition rate must be evaluated. For a single first-order reaction, the maximum decomposition rate is described by Equation [13]:
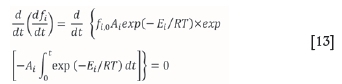
Equation [13] can be expressed as Equation [14] at a constant heating rate:
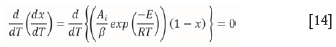
since 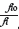 = x, for a single reaction taking place during a conversion. The determined conversion value (called xmax) is then used to calculate the corresponding Ψimax. At this point, Aí can be evaluated using Equation [15]:
= x, for a single reaction taking place during a conversion. The determined conversion value (called xmax) is then used to calculate the corresponding Ψimax. At this point, Aí can be evaluated using Equation [15]:
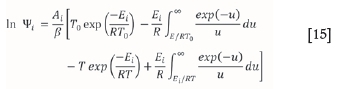
To demonstrate, for a first-order reaction, a value of x=1-e-1 ratifies Equation [14]. Then, if the first-order reaction model is expressed in terms of Ψ:

To adapt this algorithm to any other reaction model, this value of conversion (x) and corresponding value of ln [(<¥j (T)] must be modified. Hence, following the same approach, the xmaxvalue for a single reaction dissociating according to a particular reaction model taking place must be evaluated. This is described by Equation [18]:
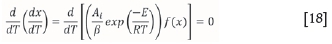
The xmaxvalues for each reaction model can be evaluated graphically or analytically for both reaction models. Equation [19] demonstrates the dependence of the Ψ expression on the reaction model.

Table I presents the results obtained upon the evaluation of Equations [18] and [19].
It is noted that the RPM reaction model has an extra parameter; this is referred to as the structural parameter φ. For the RPM, analytical evaluation of Equation [18] results in Equation [20]:
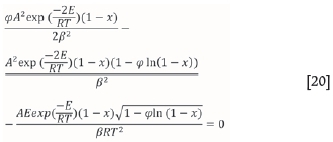
where φ is a dimensionless structural parameter in the RPM, A (s-1.m-1) is the grouped pre-exponential factor, and β is the constant heating rate (K/s). The structural parameter φ is calculated for all simulations using values applicable to common porous solids (Bhatia and Perlmutter, 1980). The values provided result in a structural parameter value of φ= 0.88. This value is used in the simulations presented in the current study.
However, when applying the algorithm to experimental data, the structural parameter is initially unknown. It is evaluated by regression for the particular data in the algorithm. It is also noted that the xmaxvalue varies with the structural parameter as described by Equation [20]. A parametric study was carried out to evaluate the sensitivity of xmaxas the structural parameter is varied at different sets of A and E. During this study, negligible changes in the xmax value were observed. It was then assumed that the value of xmaxat 0.6502 was applicable as a fixed point of maximum rate of dissociation for the RPM.
Equation [20] cannot be expressed explicitly in terms of x. An error minimization method can be applied, with a series of proposed values of E, A, and φ to determine x at which the ith reaction (not the overall material reaction) reaches a maximum rate of decomposition. This value is substituted into the manipulated RPM expression in order to determine ln[(Ψi(T)]. The modification of the original algorithm (Scott et al., 2006a) is needed in the evaluation of Ψ.
Following each fractional conversion, a set of Eiand Ai values can be determined. At this stage the algorithm has departed from iso-conversion methods in that it has determined the pre-exponential factor values without the need for an entirely separate method.
Lastly, the mass fractions must be determined. This step is vital since it further distinguishes the algorithm from other common methods. The mass fraction values allocated to each fractional conversion help identify which reactions are actually happening, and which reactions are spurious (Scott et al., 2006a). This result will produce discreet kinetic parameter values rather than a range of values which would otherwise have to be averaged, thus compromising accuracy. Now Equation [7] can be written in matrix form as M = Ψf such that:
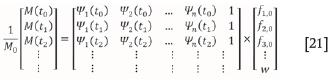
This form is used in the final step. The fiovalues for each component i are calculated using linear least squares. Equation [21] is used by specifying an E and A value and generating the candidate reactions to populate the matrix Ψ. The Mvector is the predetermined mass loss profile of the simulated or experimental reaction. Note that resulting matrix Ψ is not a square matrix. Therefore, when thef vector is calculated using matrix inversion such that M · ψ-1=f there is no exact solution. The least squares (non-negative constraint) function in Matlab (lsqnonneg is therefore used to numerically determine the f0elements in the fvector that satisfy M · Ψ-1=f.
As derived in Table I, Equation [22] is used for the evaluation of the matrix Ψ for the RPM.
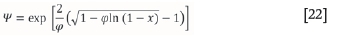
The evaluated matrix is then used in the lsqnonneg algorithm in Matlab® to do the matrix inversion and generate the vector f. The Eiand Aivalues corresponding to non-zero fi value(s) are the kinetic parameters considered to represent the reaction(s) occurring in the fuel mass.
Results and discussion
The modified algorithm was tested on two different sets of data. Following the methodology of Scott et al. (2006a), the algorithm was tested on (1) simulated conversion data, and (2) experimental thermogravimetric measurements obtained from the decomposition of a South African coal char and a 50:50 (wt%) coal-biomass blend char under CO2 atmosphere. Using Matlab software and Equation [22], typical thermogravimetric data of a reaction progressing according to a particular reaction model at a specified heating rate and kinetic parameters was generated. The algorithm was then applied to the simulated data; the kinetic parameters determined by the algorithm were compared to the kinetic parameters specified for the simulation of the data as a measure of accuracy.
Application to simulated conversion data
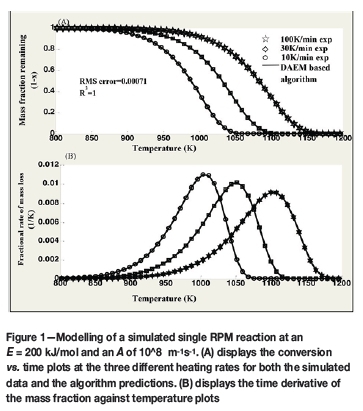
Test 1: Single RPM reaction
A single RPM reaction was simulated at three different heating rates: 10, 30, and 100 K/min. The first two heating rates were used for determining the kinetic parameters, whereas the third heating rate was merely used to show how the kinetic parameters derived by the algorithm extrapolate to different heating rates (Scott et al., 2006a). Figures 1 and 2 display the results obtained. The error values reported are the correlation coefficient (R2) and the root mean square (RMS) error value. The correlation coefficient is defined by Equation [23] (Draper and Smith, 1981).

Here, y is the model-predicted dependant variable, y{is the experimentally determined dependant variable, and y is the mean of the experimental values. The RMS error value is defined as:

As observed in Figure 1, the model predicts the simulated data to accuracies with a R2 error of 1, and a corresponding RMS error of 0.00071. For this reaction, an E of 200 kJ/mol and an A of 108 m-1s-1 is prescribed for the simulation.
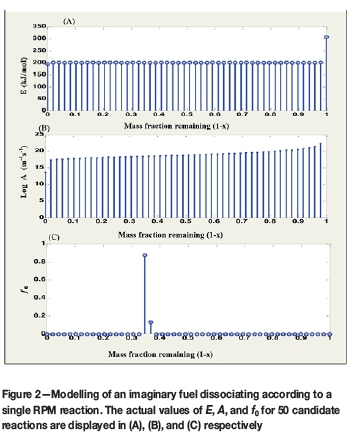
Figure 2 presents the actual kinetic parameters evaluated by the model for 50 candidate reactions; i.e 50 unique conversion points. As noted by Scott et al. (2006a), the mass fractions allocated are zero for all the other reactions except for the two points straddling the value of x = 0.6502. This is the value at which the reaction is assumed to be dominating.
Test 2: Seven RPM reactions
Seven RPM reactions were in turn simulated at the three heating rates. The kinetic parameters used in the simulation are displayed in Table II. The simulated data was applied on to the algorithm and Figures 3 and 4 display the results obtained.
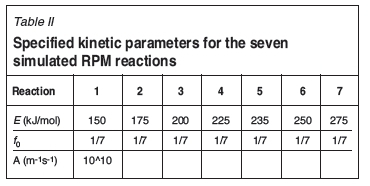
Once more, overlapping parallel reactions of Evalues of 225, 235, and 250 kJ/mol, were selected.
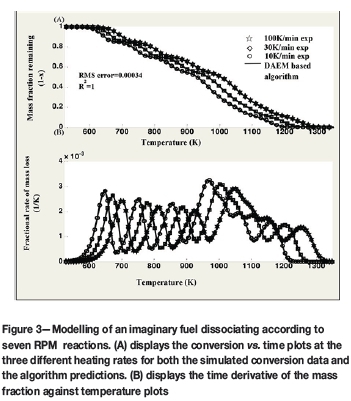
As observed in Figure 3, the kinetic parameters determined by the algorithm predict the simulated data, even at the 100 K/min heating rate, which was not used for kinetics determination. A RMS error value of 0.00034 and R2value of 1 are reported.
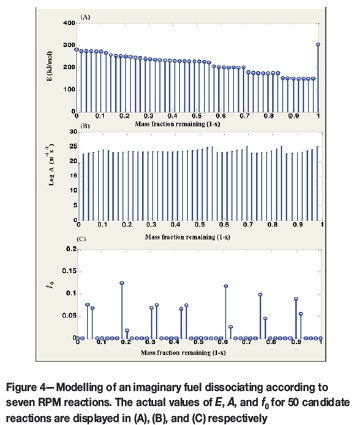
Figure 4 displays the actual kinetic parameters evaluated by the algorithm for 50 candidate reactions. Once more, the mass fractions are allocated to the actual reactions taking place at the point of dominance. At this point it can be concluded that the algorithm presented by Scott et al. (2006a), for determining the kinetic parameters of a complex fuel dissociating under the first-order reaction model has been successfully adapted to RPM behaviour.
Figure 5 presents the general steps for the adaptation of the algorithm to other reaction models.

Application of the DAEM-based algorithm to thermogravimetric analysis
Thermogravimetric analysis was carried out on the decomposition of a South African coal char and a 50:50 (wt%) coal and biomass (pinewood chips) blend under CO2 atmosphere. The samples were characterized by the use of proximate analysis as given in Table III.
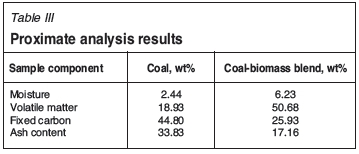
A significant increase in moisture and volatile matter is observed in the coal-biomass blend when compared to the plain coal proximate analysis, as illustrated by Table III. A decrease in the fixed carbon and ash content is also observed.
A TA Instruments SDT-Q600 high-temperature thermogravimetric analyser (TGA) was used for the experiments. Samples with nominal diameters up to 150 μm were used to produce the chars via thermogravimetry. The samples were heated at 20 K/min to 1623 K and then held at 1023 K for 15 minutes to ensure all volatiles were driven off. The coal char samples were heated at three heating rates, 12, 15, and 20 K/min, from ambient temperature to 1573 K. A constant air flow rate of 65 ml/min CO2 and 5 ml/min N2 was maintained throughout the analysis. The measurements were applied to the algorithm, and the outcome is presented in Figures 6 and 7. When dealing with experimental data, the structural parameter (φ) is not known. While it can be measured experimentally (Su and Perlmutter, 1985), other studies (Everson et al., 2006; Bhatia and Vartak, 1996) suggest that it is best to find the structural parameter via regression analysis to avoid the need for lengthy experimental procedures, as well to ensure improved accuracy and reliability of the kinetic parameters. This was carried out using an error minimizing regression method presented by Sangtong-Ngam and Narasingha (2008). This approach is characterized by Equation [25]:

where err1is the error between the calculated and experimental data at point 1, errnis the error between the calculated and experimental data at point n, and n is the number of points in the data-set. The fminbnd algorithm in Matlab (Matlab R14, The Mathworks Inc.) is used to vary the value of φ within a predetermined range until σ is minimized. This is a further modification applied to the original algorithm which is necessary for processing RPM data.
As presented in Figure 6, the algorithm results demonstrate excellent agreement with the experimental results obtained. RMS error values of 0.0043 and 0.0050 were determined for the plain coal char and coal-biomass blend char respectively. Corresponding R2 values of 0.9999 and 0.9998 are reported for the two chars as illustrated by Figure 6. The actual kinetics obtained are as outlined in Table IV. The RPM was suitable for the modelling of the conversion of both chars.
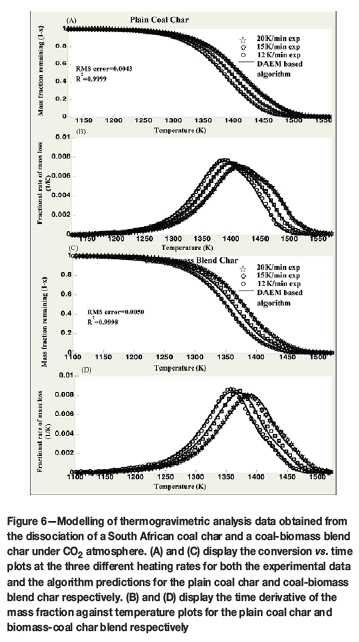
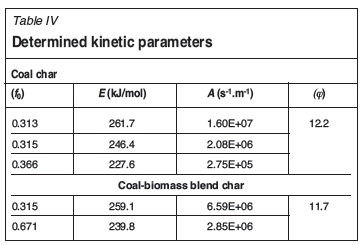
Table IV illustrates changes in the kinetic parameters of the Boudouard reaction for both chars. The different sets of kinetic parameters determined during the reaction may be aligned to the limited predicting capabilities of the RPM, such as 'catalytic effects, initially closed porosity, inhomogeneities of the active surface area, incomplete pore utilization and changes in concentrations of active sites with conversion' (Smith et al., 1994) among others. However, the variation in kinetic parameters has also been explained in the literature as due to the shifting of the reaction from the chemical reaction-controlled regime towards the pore diffusion-controlled regime (Babinski et al., 2013). This was indicated by the decrease in E from the beginning of the reaction towards its completion (Lahijani, Zainal, and Mohamed, 2012; Mani, Mahinpey, and Murugan, 2011; Hurt and Calo, 2001). The ratio of the initial E to the final E is only 0.87 and 0.93 for the plain coal char and the coal-biomass blend char respectively, instead of 0.5 for a complete shift into the diffusion-controlled regime (Babinski et al., 2013). This indicates that the reaction is predominantly taking place under the chemical reaction-controlled regime. Table IV also presents three sets of kinetic parameters for the plain coal char conversion and two sets of kinetic parameters determined during the conversion of the coal and biomass char blend. The different number of reactions/kinetic parameter sets identified may indicate the presence of synergy between the two materials during gasification. Unlike most iso-conversion methods, e.g. Vyazovkin and Friedmen methods (Babinski et al., 2003), which evaluate the reaction kinetics at a particular conversion, the algorithm was able to provide the specific mass fraction of the reactive material dissociating under a particular set of kinetic parameters. This provides more detailed kinetic information which could potentially improve the application of kinetic parameters in the design and optimization of conversion systems.
The actual Evalues determined by the algorithm for both the pure coal char and the coal and biomass char blend were found comparable to values reported by Zhu, Sing, and Lin (2008) and Liu et al. (2000) for the gasification of coal chars. Table IV also displays the determined φ values of 12.2 and 11.7 for the two chars, which compare to the range of values of 2.2 to 50 reported in the literature (Liu and Niksa, 2004; Zolin, 2001; Charpenay, Serio, and Solomon, 1992. A single φ for both chars was sufficient to describe the reaction progression despite the changes observed in the rest of the kinetic parameters.
It is essential to define the kinetic parameters in context prior to comparison. Ehas been described as the minimum amount of energy required to start a chemical reaction (Vittee, 2012; Brown, LeMay, and Bursten, 2009). Lower E reactions have therefore been interpreted to imply that the reaction takes place more spontaneously, or that the conditions and reactants are more reactive compared to reactions of higher E. The pre-exponential factor, also termed the frequency factor, indicates the number of collisions of particles occurring with the correct orientation and sufficient energy to enable a reaction to take place (Brown, LeMay, and Bursten, 2009). A lower E and higher pre-exponential factor relates to a higher degree of reactivity, whereas a higher E and lower pre-exponential factor indicates a lower degree of reactivity.
When comparing the kinetics of the plain coal char and that of the coal-biomass blend, the differences are not quite palpable. When simply considering the average Eand A, the blend would be interpreted to have less reactive kinetics. However, the unique feature of the DAEM allows for a more detailed analysis and comparison of the obtained kinetics by determining the mass fractions associated with each reaction taking place.
The blend presents an Erange between 239.8 kJ/mol and 259.1 kJ/mol and corresponding grouped pre-exponential factors in the range of 2.85E+6 to 6.59E+6. The lower E describes the dissociation of the majority of the reactive material (67%). The plain coal char is described by an E range of 227.6 kJ/mol to 261.6 kJ/mol, and a corresponding A range of 2.75E+5 to 1.6E+7 attributed to three virtually equal mass fractions. The relatively high frequency factor (1.6E+7), requires a high activation energy (261.6 kJ/mol) for the first fraction; this reaction is comparable to the higher E (259.1 kJ/mol) reaction in the blend. The second mass fraction is described by an E of 246.4 kJ/mol and A of 2.08E+6, both parameters implying a slightly less reactive profile than those of the major components of the blend. The last mass fraction component of the pure coal char presents a lower E of 227.6 kJ/mol and significantly lower A of 2.75E+5, making the reaction profile somewhat comparable to that of the major mass fraction component in the blend ( E = 239.8 kJ/mol and A = 2.85E+6). Hence, a complete analysis of the kinetics, considering the mass fraction components, shows an improvement in reactivity in the coal-biomass blend char compared to the plain coal char. It is also clear that the φ value has been reduced slightly from the pure coal char to the coal and biomass char blend. As described by Bhatia and Perlmutter (1980), two opposing forces contribute to surface area growth; pore expansion and pore collapse due to coalescence. A φ greater than 2 has been determined to indicate the prevalence of pore growth, whereas that lower than 2 is believed to demonstrate higher pore coalescence (Bhatia and Perlmutter, 1980). Since the two φ values already indicate significant pore growth, the lower φ value is assumed to indicate a slightly higher prevalence of pore coalescence; this is considered to be a possible contributing factor to the improved reactivity of the blended chars.
Conclusion
The DAEM-based model developed by Scott et al. (2006a) has been successfully modified for the dissociation of complex fuels according to the RPM. Despite the derivation of the DAEM-based algorithm being based upon first-order reaction kinetics, a general method to adapt the algorithm to other reaction models has been outlined. The DAEM-based algorithm has also been shown to be model-independent in the determination of E.
The modified algorithm has been tested on simulated conversion data for a typical fuel dissociating according to
> Single RPM reactions
> Seven RPM reactions, each with a specified E and A.
The modified DAEM-based algorithm was also applied to experimental data obtained from the conversion of a South African coal char and a coal-biomass blend char via thermogravimetry in a CO2 atmosphere. The algorithm evaluated E, A, and fvalues for the number of reactions taking place during the tests. The kinetic parameters determined by the algorithm predicted the simulated and experimental data to high accuracies with R2 values of 1 to 0.9998, and RMS error values of 0.00034 to 0.0050.
The algorithm also demonstrates the ability to determine any changes in the kinetic parameters occurring during the reaction; this provides more insight to the reaction mechanisms taking place during the conversion.
By determining the reactive mass fraction component that is reacting according to a particular set of kinetic parameters, the DAEM-based algorithm provides unique and accurate kinetic details for the modelling of conversion processes. The value of the mass transfer component was demonstrated in comparing kinetic parameters of different char blends. The mass fraction components provide a higher degree of detail, which is valuable as it enhances the understanding, analysis, and comparison of the reaction profiles of complex fuels. The addition of biomass to the coal char was shown to improve the overall reactivity of the coal. A reduction in the number of reactions taking place during the conversion of the pure coal char and biomass/coal blend was observed, indicating the possibility of synergetic behaviour between the blended char components. A reduction in the structural parameter was also observed. The determined kinetic parameters are comparable to values reported in the literature.
Notation
Ao Pre-exponential factor (s-1)
A Grouped pre-exponential factor (s-1m-1)
b Dimensionless constant
Cg Concentration of gaseous reactant (mol.m-3)
E Activation energy (J.mol-1)
fi Mass fraction of material
Mv( t) Total mass of volatile matter at time t, (kg)
M(t) Total mass remaining at time t, (kg)
M0 Initial sample mass (kg)
R Ideal gas constant (kJ.mol-1K-1)
R2 Coefficient of linear correlation
T Temperature (K)
T0 Initial temperature (K)
μ Dummy variable
x0 Initial char mass fraction
x Mass fraction of char at a given time during conversion
yiExperimentally determined dependent variable
yi Model-predicted dependent variable
yi Mean of experimentally determined dependent variables
Subscripts
i indicates reaction number, or component number
v indicates property of volatile matter
0 indicates initial state
Greek letters
a Fractional conversion
β Heating rate (K.s-1)
φ Structural [parameter
Ψ exp [A (E) ƒ exp((-E)/RT)dt] (reaction function) 0
References
Babinski, P., Labojko, G., Kotyczka-Moran ' ska, M., and Plis, A. 2013. Kinetics of coal and char oxycombustion studied by TG-FTIR. Journal of Thermal Analysis and Calorimetry, vol. 113, no. 1. pp. 371-378. DOI 10.1007/s10973-013-3002-x [ Links ]
Bhatia, S.K. and Vartak, B.J. 1996. Reaction of microporous solids: The discrete random pore model. Carbon, vol. 34, no. 11. pp. 1383-1391. [ Links ]
Bhatia, S.K. and Perlmutter, D.D. 1980. A random pore model for fluid-solid reactions: I. Isothermal, kinetic control. AIChE Journal, vol. 26. pp. 379-386. [ Links ]
Braun, R.L. and Burnham, A.K. 1987. Analysis of chemical reaction kinetics using a distribution of activation energies and simpler models. Energy and Fuels, vol. 1, no. 2. pp. 153-161. [ Links ]
Brown, T., LeMay, H., and Bursten, B. 2009. Chemistry: The Central Science. International edition. Pearson Prentice Hall. [ Links ]
Charpenay, S., Serio, M.A., and Solomon, P.R. 1992. The prediction of coal char reactivity under combustion conditions. Twenty-Fourth Symposium (International) on Combustion/The Combustion Institute. pp. 1189-1197. [ Links ]
Draper, N.R.W. and Smith, H. 1981. Applied Regression Analysis. 2nd edn. Wiley, New York. [ Links ]
Everson, R.C., Neomagus, H.W.J.P., Kasaini, H., and Njapha, D. 2006. Reaction kinetics of pulverized coal-chars derived from inertinite-rich coal discards: Gasification with carbon dioxide and steam. Fuel, vol. 85, no. 7-8. pp. 1076-1082. [ Links ]
Hurt, R.H. and Calo, J.M. 2001. Semi-global intrinsic kinetics for char combustion modeling. Combustion and Flame, vol. 125, no. 3. pp. 1138-1149. [ Links ]
Irfan, M.F., Usman, M.R., and Kusakabe, K. 2011. Coal gasification in CO2 atmosphere and its kinetics since 1948: a brief review. Energy, vol. 36, no. 1. pp. 12-40. [ Links ]
Kirtania, K. and Bhattacharya, S. 2012. Application of the distributed activation energy model to the kinetic study of pyrolysis of the fresh water algae Chlorococcum humicola. Bioresource Technology, vol. 107. pp. 476-487. [ Links ]
Lahijani, P., Zainal, Z.A., and Mohamed, A.R. 2012. Catalytic effect of iron species on CO2 gasification reactivity of oil palm shell char. Thermochimica Acta, vol. 546. pp. 24-31. [ Links ]
Liu, G. and Niksa, S. 2004. Coal conversion submodels for design applications at elevated pressures. Part II. Char gasification. Progress in Energy and Combustion Science, vol. 30, no. 6. pp. 679-717. [ Links ]
Liu, G., Tate, A.G., Bryant, G.W., and Wall, T.F. 2000. Mathematical modeling of coal char reactivity with Co2 at high pressures and temperatures. Fuel, vol. 79. pp. 1145-1154. [ Links ]
Maki, T., Takatsuno, A., and Miura, K. 1997. Analysis of pyrolysis reactions of various coals including argonne premium coals using a new distributed activation energy model. Energy and Fuels, vol. 11. pp. 972-977. [ Links ]
Mani, T., Mahinpey, N., and Murugan, P. 2011. Reaction kinetics and mass transfer studies of biomass char gasification with CO2. Chemical Engineering Science, vol. 66, no. 1. pp. 36-41. [ Links ]
Miura, K. 1995. A new and simple method to estimatef(E) and k0 (E) in the distributed activation energy model from three sets of experimental data. Energy and Fuels, vol. 9. pp. 302-307. [ Links ]
Miura, K. anD Maki, T. 1998. A simple method for estimatingf(E) and k0 (E) in the distributed activation energy model. Energy and Fuels, vol. 12. pp 864-869. [ Links ]
Pitt, G.J. 1962. The kinetics of the evolution of volatile products from coal. Fuel, vol. 41. pp. 264-274. [ Links ]
Please, C.P., McGuinness, M.J., and McElwain, D.L.S. 2003. Approximations to the distributed activation energy model for the pyrolysis of various coals. Combustion and Flame, vol. 133. pp. 107-117. [ Links ]
Research and Markets. 2006. Market Analysis - Gasification of Coal and its importance in the Power Sector. [ Links ]
Rostami, A.A., Hajalgol, M.R., and Wrenn, S.E. 2004. A biomass pyrolysis sub-model for CFD applications. Fuel, vol. 83. pp. 1519-1525. [ Links ]
Rotaru, A. and Goêa, M., 2009. Computational thermal and kinetic analysis: Complete standard procedure to evaluate the kinetic triplet form non-isothermal data. Journal of Thermal Analysis and Calorimetry, vol. 97, no. 2. pp. 421-426. [ Links ]
Sangtong-Ngam, K. and Narasingha, M.H. 2008. Kinetic study of Thai-lignite char gasification using the random pore model. Thammasat International Journal of Science and Technology, vol. 13. pp. 16-26. [ Links ]
Scott, S.A., Dennis, J.S., Davidson, J.F., and Hayhurst, A.N. 2006a. An algorithm for determining the kinetics of devolatilisation of complex solid fuels from thermo-gravimetric experiments. Chemical Engineering Science, vol. 61, no. 8. pp. 2339-2348. [ Links ]
Scott, S.A., Dennis, J.S., Davidson, J.F., and Hayhurst, A.N. 2006b. Thermogravimetric measurements of the kinetics of pyrolysis of dried sewage sludge. Fuel, vol. 85, no. 9. pp. 1248-1253. [ Links ]
Smith, K.L., Smoot, L.D., Fletcher, T.H., and Pugmire, R.J. 1994. The Structure and Reaction Processes of Coal. Plenus Chemical Engineering Series. 387 pp. [ Links ]
Starink, M.J. 2003. The determination of activation energy from linear heating rate experiments : a comparison of the accuracy of iso-conversion methods. Thermochimica Acta, vol. 404, no. 1-2. pp.163-176. [ Links ]
Su, J.L. and Perlmutter, D. 1985. Effect of pore structure on char oxidation kinetics. AIChE Journal, vol. 31. pp. 973-981. [ Links ]
Vittee, T. 2012. Determination of Kinetics of Char Reactivity with Carbon Dioxide using Thermogravimetry and the Distributed Activation Energy Model. MSc dissertation, University of the Witwatersrand, Johannesburg, South Africa. [ Links ]
Vyazovkin, S. 2008. Isoconversional kinetics. Handbook of Thermal Analysis and Calorimetry. Brown, M.E. and Gallagher, P.K. (eds.). Elsevier. Chapter 13, pp. 503-537. [ Links ]
Zhu, W., Song, W., and Lin, W. 2008. Catalytic gasification of char from co-pyrolysis of coal and biomass. Fuel Processing Technology, vol. 89, no. 9. pp. 890-896. [ Links ]
Zolin, A. 2001. Reactivity of Solid Fuels. PhD dissertation, Department of Chemical Engineering, Technical University of Denmark, Denmark. [ Links ]
Paper received Apr. 2015
Revised paper received Jun. 2015
©The Southern African Institute of Mining and Metallurgy, 2016. ISSN2225-6253.
1 integral iso-conversion methods that do not make an approximation to the temperature integral are known as 'algorithmic methods', but are generally considered highly calculation-intensive.


{kind=link}