










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkJournal of the Southern African Institute of Mining and Metallurgy
On-line version ISSN 2411-9717
Print version ISSN 2225-6253
J. S. Afr. Inst. Min. Metall. vol.115 n.10 Johannesburg Oct. 2015
http://dx.doi.org/10.17159/2411-9717/2015/v115n10a9
A redetermination of the structure of tetraethylammonium mer-oxidotrichlorido(thenoyltrifluoroacetyl acetonat0-K2-0,0')niobate(V)
R. Koen; A. Roodt; H. Visser
Department of Chemistry, University of the Free State, South Africa
SYNOPSIS
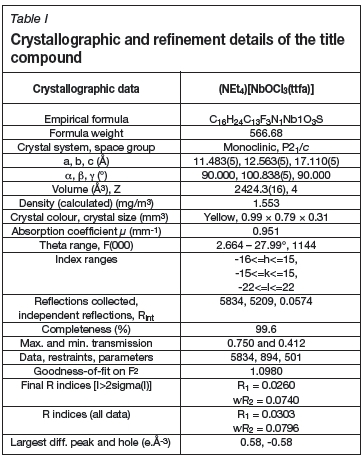
The tetraethylammonium salt of the mono-anionic coordination compound me/--oxidotrichlorido(thenoyltrifluoroacetylacetonato-K2O,O')niobate(V) (NEt4)[NbOCl3](ttfa)], has been prepared under aerobic conditions and characterized by single-crystal X-ray diffraction. (NEt4)[NbOCl3](ttfa)] crystallized in the monoclinic .P2i/c space group, with a = 11.483 (5), b = 12.563 (5), c = 17.110(5) Â, and β = 100.838 (5)°. The complex structure exists in a 50.0% (NbA) : 50.0% (NbB) positional disorder ratio.
Keywords: Bidentate, niobium(V), disorder.
Introduction
Complexes containing organometallic type β-diketone ligands with O,O and O,N donor atoms are used widely in coordination chemistry and have applications in catalysis, radiopharmaceuticals, etc. (Schutte et al., 2011; Roodt, Visser and Brink, 2011; Brink et al., 2010; Otto et al., 1998). These ligand systems are very useful because of their highly coordinative nature, high solubility, and also due to their ability to be functionalized with various substituents on the carbonyl carbon atoms (Schutte et al., 2011).
Only a small number of β-diketonate ligands have successfully been coordinated to a Nb(V) metal centre, with only a select few being characterized by X-ray crystallography (Viljoen, 2009; Bullen, Mason and Pauling, 1965; Preuss, Lamding and Mueller-Becker, 1994; Funk, 1934; Davies, Leedlam and Jones, 1999; Allen, 2002. The synthesis and crystal structure determination at room temperature of mer-oxidotrichlorido (thenoyltrifluoroacety-lacetonato-s2O,O')niobate(V) {(NEt4)[NbOCl3(ttfa)]} was first reported by Daran et al. in 1979. Accordingly, for this current investigation, (NEt4)[NbOCl3(ttfa)] was re-evaluated at 100 K to determine if the structural features might change with temperature.
This study of this structure forms part of an AMI-funded programme to better understand the solid-state characteristics of Ta(V) and Nb(V) complexes and the influences of the electron-donating and -withdrawing groups of the β-diketone on the activity induced and reaction mechanisms at these metal centres.
Experimental
Materials and instruments
All chemicals used for the synthesis and preparation of the complexes were of analytical grade and were purchased from Sigma- Aldrich, South Africa.
The 1H-, 13C-, and 19F FT-NMR solution-state spectra were recorded on a Bruker AVANCE II 600 MHz (1H: 600.28 MHz; 13C: 150.96 MHz; 19F: 564.83 MHz) or Bruker DPX 300 MHz (1H: 300.13 MHz; 13C: 75.47 MHz; 19F: 282.40 MHz) nuclear magnetic resonance spectrometer using the appropriate deuterated solvent. Chemical shifts, δ, are reported in ppm. 1H NMR spectra were referenced internally using residual protons in the deuterated solvents, Acetonitrile-d3 [CD3CN = 1.94(5) ppm]. 13C NMR spectra were similarly referenced internally to the solvent resonance [CD3CN = 1.39(4) ppm and 118.69(8) ppm] with values reported relative to tetramethyl-silane (δ 0.0 ppm).
The X-ray intensity data was collected on a Bruker X8 ApexII 4K Kappa CCD area detector diffractometer, equipped with a graphite monochromator and MoKa fine-focus sealed tube (λ = 0.71069 Â, T = 100(2) K) operated at 2.0 kW (50 kV, 40 mA). The initial unit cell determinations and data collection were done by the SMART (Bruker, 1998a) software package. The collected frames were integrated using a narrow-frame integration algorithm and reduced with the Bruker SAINT-Plus and XPREP software package (Bruker, 1999) respectively. Analysis of the data showed no significant decay during the data collection. The data was corrected for absorption effects using the multi-scan technique SADABS Bruker, 1998b) and the structure was solved by the direct methods package SIR97 (Altomare et al., 1999) and refined using the WinGX (Farrugia, 1999) software incorporating SHELXL (Sheldrick, 1997). The final anisotropic full-matrix least-squares refinement was done on F2. The aromatic protons were placed in geometrically idealized positions (C-H = 0.93 - 0.98 À) and constrained to ride on their parent atoms with Uiso(H) = 1.2Ueq(C). Non-hydrogen atoms were refined with anisotropic displacement parameters. The graphics were obtained with the DIAMOND program (Brandenburg, 2006) with 50% probability ellipsoids for all non-hydrogen atoms.
Synthesis of (NET)4([NbOCl3(ttfa)]( 1)
(Et4N)[NbCl6] (0.500 g, 1.147 mmol) was added to 4,4,4-trifluoro-/(2-thienyl)-/,3-butanedione (ttfaH) (0.327 g, 1.147 mmol) in acetonitrile (20 cm3). The resulting solution was heated to 50°C and stirred for 30 minutes. The excess solvent was evaporated and dark yellow plate-like crystals of the title compound (1), suitable for X-ray diffraction, were obtained (0.565 g, yield 89 %). IR (ATR, cm-1): v(Nb=O)= 952. 1H NMR (300.13 MHz, Acetonitrile-rf3, ppm): δ = 5.88 (s, 1H), 6.83 (m, 1H), 6.93 (dd, 1H), 7.40 (dd, 1H). 13C NMR (75.47 MHz, Acetonitrile-<4 ppm): δ= 30.1, 118.8, 123.9, 130.0, 137.4, 142.0, 182.3. 19F NMR (564.83 MHz, Acetonitrile-rf3, ppm): -73.37.
Results and discussion
The title compound was previously prepared by Daran et al. (1979), with X-ray diffraction data collected at room temperature. For this study, the reaction was modified as described above and the data collected at 100 K.
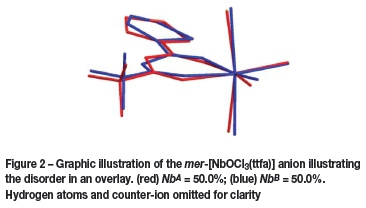
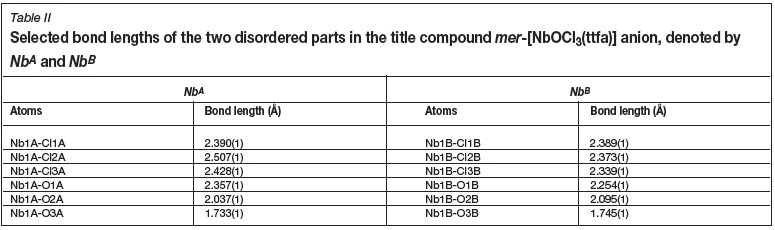
The compound crystallizes in the monoclinic space group, P21/c, with four molecules in the unit cell (Z = 4). The asymmetric unit consists of a Nb(V) metal centre surrounded by three crystallographically independent chlorido groups (Cl1A - Cl3A), an oxido (O3A) and one 0,<7-bonded thenoyl-trifluoroacetonato ligand and a tetraethylammonium cation. A graphic illustration is shown in Figure 1. The complex molecule and the counter-ion are disordered over two positions in a 50 NbA 50 NbB ratio as shown in Figure 2. General crystallo-graphic details are presented in Table I, while selected bond lengths and bond angles are listed in Tables II and III respectively.
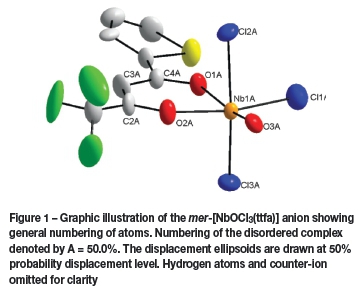
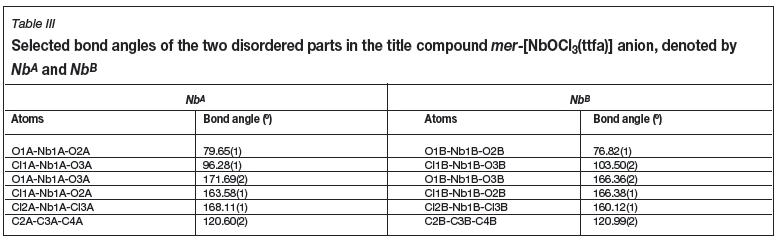
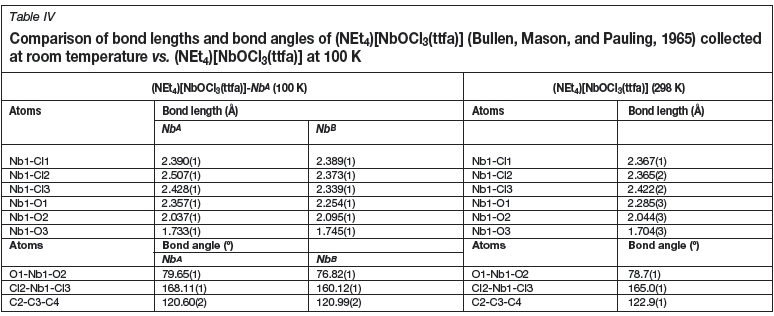
A distorted octahedral geometry is displayed for NbA and NbB. The Nb-Claxial distances for NbA vary between 2.428(1) and 2.507(1) À and the Nb1A-Cl1A and Nb1A-O3A have distances of 2.390(1) and 1.733(1) Å, respectively. When comparing Nb1A-O1A and Nb1A-O2A bond lengths, distances of 2.357(1) vs. 2.037(1) À are observed. This difference is due to the effects of the electron withdrawing, CF3 substituent on the bidentate ligand backbone causing a longer NbA-O1A bond length. The trans Cl2-Nb1-Cl3 angle is 168.11(1)°, while the O1-Nb1-O2 bite angle is 79.65(1)°. A similar distortion is observed for NbB, with bond- lengths and angles in accordance with NbA.
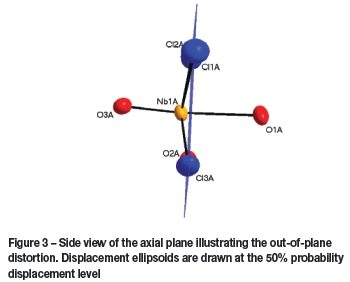
The coordination plane constructed through Cl1A, Cl2A, Cl3A, and O2A, as indicated in Figure 3, shows the niobium metal centre is slightly shifted out of this plane by 0.2751(3) Å.
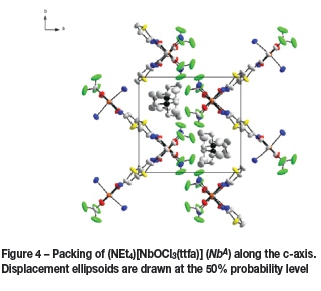
The molecular packing within the unit cell is illustrated in Figure 4. The packing illustrates a 'head-to-tail' arrangement when viewed along the c-axis. There are no classical hydrogen bonds or interactions observed in this structure.
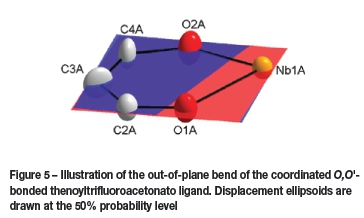
In Figure 5, two coordination planes are illustrated; the first one constructed through O1A, O2A, and Nb1A, and the second through O1A, C2A, C3A, C4A, and O2. The angle between planes revealed the slight, 1.677° out-of-plane bend of the coordinated Ο,Ο'-bonded thenoyltrifluoroacetonato ligand. This also contributes to the distorted octahedral geometry.
The crystal structure determination of the published complex was performed at room temperature (298 K), while the synthesized analogue (1) was determined at 100(2) K. Data obtained for the title compound (1) correlates well with the previously published structure (Daran et al. 1979). The disordered part denoted by NbBdiffers less from the published structure and is probably a better representation of the anionic complex. Table IV illustrates a comparison between bond angles and distances of the published structure vs. the structure collected at 100K. The greatest difference between the two complexes is the positional disorder observed in the newly synthesized product.
Conclusions
A simplified method to obtain (NEt4)[NbOCl3(ttfa)] in aerobic conditions is reported. This highlights the misrepresentation of the 'extreme sensitivity' of niobium(V) complexes to air and water. Clearly, the exclusion of oxygen is not that important, while the exclusion of water is, since it will increase hydrolysis and thus the loss of chloride in favour of aqua, hydroxide, or oxo coordination. The crystallographic investigation revealed that this structure exhibited a 50:50 positional disorder. The electron withdrawing effects of the CF3 substituent on the bidentate ligand backbone is illustrated by the longer Nb-O bonds of Nb1A-O1A and Nb1B-O1B vs. Nb1A-O2A and Nb1B-O2B. All bond lengths and angles of the complex were found to be in accordance with similar structures in the literature.
Acknowledgements
Financial assistance from the Advanced Metals Initiative (AMI) of the Department of Science and Technology (DST) of South Africa, through the New Metals Development Network (NMDN) managed by the South African Nuclear Energy Corporation Limited (Necsa) is gratefully acknowledged. Gratitude is also expressed towards SASOL, PETLabs Pharmaceuticals, and the University of the Free State for financial support of this research initiative outputs. Furthermore, this work is based on research supported in part by the National Research Foundation of South Africa (UIDs 71836 and 84913).
References
Allen, F.H. 2002. Cambridge Structural Database (CSD) Version 5.35, November 2013 update. Acta Ciystallographica, vol. B58. pp. 380-388. [ Links ]
Altomare, A., Burla, M.C., Camalli, M., Cascarano, G.L., Giacovazzo, C., Guagliardi, A., Moliterni, A.G.G., Polidori, G., and Spagna, R. 1999. SIR97: a new tool for crystal structure determination and refinement Journal of Applied Crystallography, vol. 32. pp. 115-119. [ Links ]
Brandenburg, K. 2006. DIAMOND, Release 3.0e. Crystal Impact GbR, Germany. [ Links ]
Brink, A., Roodt, A., Steyl, G., and Visser, H.G. 2010. Steric vs. electronic anomaly observed from iodomethane oxidative addition to tertiary phosphine modified rhodium(I) acetylacetonato complexes following progressive phenyl replacement by cyclohexyl [PR3 = PPh3, PPh2Cy, PPhCy2 and PCy3]. Dalton Transactions, vol. 39. pp. 5572-5578. [ Links ]
Bruker AXS Inc. 1998a. Bruker SMART-NT Version 5.050, Area-Detector Software Package. Madison, WI, USA. [ Links ]
Bruker AXS Inc. 1998b. Bruker SADABS Version 2004/1, Area Detector Absorption Correction Software. Madison, WI, USA. [ Links ]
Bruker AXS Inc. 1999. Bruker SAINT-Plus Version 6.02 (including XPREP),. Area-Detector Integration Software. Madison, WI, USA. [ Links ]
Bullen, G.J., Mason, R., and Pauling, P. 1965. The crystal and molecular structure of bis(acetylacetonato)nickel (II). Inorganic Chemistry, vol. 4. pp. 456-462. [ Links ]
Daran, J., Jeanin, Y., Guerchais, J.E., and Kergoat, R. 1979. The crystal structure of tetraethylammonium trichlorooxo(1,1,1-trifluoro-4-thenoyl- 2,4-butanedionato)niobate(V). Inorganica ChimicaActa, vol. 33. pp. 81-86. [ Links ]
Davies, H.O., Leedlam, T.J., and Jones, A.C. 1999. Some tantalum(V) β- diketonate and tantalum(V) aminoalcoholate derivatives potentially important in the deposition of tantalum-containing materials. Polyhedron, vol. 18. pp. 3165-3174. [ Links ]
Farrugia, L.J. 1999. WinGX suite for small-molecule single-crystal crystallography. Journal of Applied Crystallography, vol. 32. pp. 837-838. [ Links ]
Funk, H. 1934. Über die einwirkung von niob- und tantalpentachlorid auf organische verbindungen (IV. Mitteil.). Berichte der Deutschen Chemischen Gesellschaft, vol. 62. pp. 1801-1808. [ Links ]
Otto, S., Roodt, A., Swarts, J.C., and Erasmus, J.C. 1998. Electron density manipulation in rhodium(I) phosphine complexes: structure of acetylacet- onatocarbonylferrocenyl diphenylphosphinerhodium(I). Polyhedron, vol. 17. pp. 2447-2453. [ Links ]
Preuss, F., Lamding, G., and Mueller-Becker, S. 1994. Oxo- und thiotan- tantal(V)-verbindungen: Syntese von TaOX, und TaSX (X = OR, SR), Z. Zeitschriftfür Anorganische und allgemeine Chemie, vol. 620. pp. 1812-1820. [ Links ]
Roodt, A., Visser, H.G., and Brink, A. 2011. Structure/reactivity relationships and mechanisms from X-ray data and spectroscopic kinetic analysis. Crystallography Reviews, vol. 17. pp. 241-280. [ Links ]
Schutte, M., Kemp, G., Visser, H.G., and Roodt, A. 2011. Tuning the reactivity in classic low-spin d(6) rhenium(I) tricarbonyl radiopharmaceutical synthon by selective bidentate ligand variation (L,L'-Bid, L,L ' = N,N', N,O & O,O' donor atom sets) infc-[Re(CO)3(L,L'-Bid)(MeOH)]n complexes. Inorganic Chemistry, vol. 50. pp. 12486-12498. [ Links ]
Sheldrick, G.M. 1997. SHELXL97. Program for crystal structure refinement. University of Göttingen, Germany. [ Links ]
Viljoen, J.A. 2009. Speciation and interconversion mechanism of mixed halo O,O'- and N,O-bidentate ligand complexes of hafnium. MSc dissertation, Universtry of the Free State. 132 pp. [ Links ]
Paper received Aug. 2015
Revised paper received Aug. 2015.


{kind=link}
{kind=link}
{kind=link}