










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkJournal of the Southern African Institute of Mining and Metallurgy
On-line version ISSN 2411-9717
Print version ISSN 2225-6253
J. S. Afr. Inst. Min. Metall. vol.113 n.8 Johannesburg Jan. 2013
CONFERENCE
SO2 - an indirect source of energy
R.J. KriekI; J.P. van RavenswaayI; M. PotgieterI; A. CalitzI; V. LatesI; M.E. BjörketunII; S. SiahrostamiII; J. RossmeislII
IPGM Group, Research Focus Areafor Chemical Resource Beneficiation (CRB), North-West University, Potchfstroom, South Africa
IICentre for Atomic-scale Materials Design (CAMD), Department of Physics, Technical University of Denmark, Denmark
ABSTRACT
Global sulphur dioxide (SO2) emissions peaked around the mid-1970s, after which they declined. However, with the growth of specifically China, emissions are on the rise again. In 2008, global anthropogenic SO2 emissions totalled 127 Mt, with energy production accounting for 63.2 Mt and metal-related processes 12.8 Mt.
As a well-known gaseous pollutant, SO2 is not per se known as a source of energy. However, in the presence of water SO2 can be electro-oxidized at the anode of an electrolyser to produce hydrogen ions, which in turn can be reduced at the cathode of the electrolyser to produce hydrogen gas. Gaseous emissions of SO2 can therefore be cleaned up with the simultaneous production of hydrogen, an energy store or carrier, which provides an economic offset to the overall cost of this potential remediation process. This process forms part of the Hybrid Sulfur (HyS) cycle as well as the once-through HyS (OTHyS) cycle.
Indications are that the greatest stride towards the development of an effective electrolyser for the electro-catalytic oxidation of SO2 requires the development of an anode electrocatalyst exhibiting enhanced activity for the electro-oxidation of SO2 A critical review will be presented on the research and development of such an anode electrocatalyst, and a strategy for a more effective research and development effort will be discussed. This will include theoretical studies on the electro-catalytic oxidation of SO2 on different metal-based catalytic surfaces (for which some preliminary results are presented) in conjunction with combinatorial (simultaneous multi-metal multi-electrode) electroche6mistry studies as well as single-electrode electrochemistry studies.
Keywords: sulphur dioxide, electro-oxidation, electrolyser, electrocatalyst.
Introduction
Sulphur dioxide (SO2) is a colourless, nonflammable gas that can be detected by taste and smell in the range of 0.001-0.003 ppm (World Bank, 1999; WBK & Associates, 2003). It is predominantly a pulmonary irritant affecting the upper respiratory tract (the nose and throat) and lungs, but it also affects the eyes and skin.
Volcanoes and volcanic vents are the biggest natural contributors to atmospheric SO2, whereas the main anthropogenic sources of SO2 include the combustion of fossil fuels and non-ferrous metal smelters (World Bank, 1999; WBK & Associates, 2003).
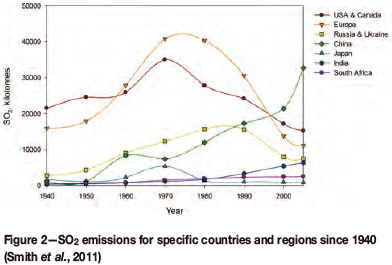
Around the mid-1970s SO2 emissions peaked, subsequently declining as a direct result of the implementation of focused abatement technologies. However, with the economic growth of specifically China, and to a lesser extent India, SO2 emissions are on the rise again (Smith et al., 2011). Spatial distribution maps drafted using data from the Emission Database for Global Atmospheric Research (EDGAR) of the European Commission's Joint Research Centre (JRC) (EC-JRC/PBL, 2010) clearly show how SO2 emissions have diminished in North America, Europe, and Japan as well as in Russia and the Ukraine, from 1970 to 2008 (Figure 1). For the same period, however, emissions have increased for China and India. According to 2005 data, China emitted in the order of 33 Mt of SO2 per annum, followed by the USA and Canada with 15 Mt, Europe with 11 Mt, India with 6.3 Mt, and South Africa with 2.5 Mt (Figure 2) (Smith et al., 2011). Approximately 90% of global anthropogenic SO2 emissions are emitted in the northern hemisphere, with coal-fired power generation being the biggest culprit. In China alone, according to 2004 figures, SO2 emitted from coal-fired power plants contributed 52.6% to the country's total annual SO2 emissions (Fang et al., 2008). This was followed by the chemical industry (10.4%), metallurgical industry (9.9%), and the building materials industry (9.2%).
SO2 can be converted in the atmosphere to sulphuric acid (H2SO4), according to the reaction steps shown in Equations [1]-[3], through a homogeneous gas-phase pathway, a homogeneous aqueous-phase pathway, or a heterogeneous pathway on the surfaces of particulate matter, or any combination of all three pathways (Bunce, 1994; Calvert and Stockwell, 1984).
S02+OH(+M) → HSO3 (M = N2 .O2 or H2O) [1]
HSO3+ O2 → SO3 + HO2 [2]
SO3 + H2O → H2SO4 [3]
A further mechanism of SO2 removal from the atmosphere is according to a catalytic pathway (Friend, 1973):
SO2 + 1/2O2 (dissolved) + H2O → H2SO4 [4]
The sulphuric acid, known as acid rain, then falls to the ground affecting vegetation, crops, natural waterways, and man-made structures.
Flue gas desulphurization (FGD) is currently the technology for removing SO2 from the exhaust flue gases of coal-fired power plants. Wet FGD technologies have been employed at the majority of international installations, with most wet limestone and lime spray installations being capable of removing about 90% of the sulphur dioxide. More than 95% sulphur dioxide removal can be achieved by employing advanced state-of-the-art wet scrubber systems (Srivastava et al., 2001). It has been reported that despite the installation of FGD technologies on some Chinese coal-fired power plants, there are no guarantees that these units were running continuously prior to 2007. Due to novel policy incentives in 2007 and the implementation of continuous emission monitoring systems, the number of Chinese coal-fired power plants fitted with FGD technologies increased from 60% in 2008 to 80% in 2010 (Su et al., 2011). However, despite the installation of SO2 abatement technologies at Chinese coal-fired power plants, the amount of SO2 being emitted is still increasing. In South Africa the Kusile coal-fired power plant, currently under construction, will be the first to have flue gas desulphurization installed.
Current abatement technologies at platinum smelters include basically two classes, i.e. those geared towards converting the sulphur dioxide into sulphuric acid, which can be used elsewhere, or those converting sulphur dioxide into a solid material that could also be used elsewhere or be disposed of. An example of such a sulphuric acid production technology from sulphur dioxide is the SULFACID® process (widely used in the pigment industry) that has been installed at Impala Platinum's smelter outside of Rustenburg, South Africa (Kruger, 2004; Westcott et al., 2007). An example of a scrubber system converting sulphur dioxide into a 'solid' material is the dual-alkali scrubber plant installed at Lonmin's smelter, which produces CaSOX, i.e. CaSO3.1/2H2O and CaSO4.2H2O (Bezuidenhout et al., 2012). Gypsum, a saleable product, could be produced if a post-oxidation step is implemented converting calcium sulphite to calcium sulphate.
These technologies, however, whether acid production or salt production, produce a material that could either be saleable or needs to be disposed of.
The Hybrid Sulfur (HyS) cycle
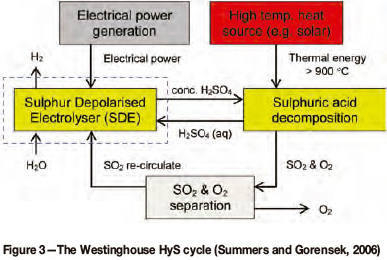
In the early 1970s Westinghouse Electric Corporation developed the HyS cycle, which is also known today as the Westinghouse cycle (Brecher and Wu, 1975). It is a thermo-chemical cycle (Figure 3) during which sulphuric acid is decomposed at approximately 900°C in a thermal decomposition reactor to produce sulphur dioxide and oxygen (Equation [5]). The sulphur dioxide, in an aqueous sulphuric acid solution, is fed to an electrolyser, also called a sulphur depolarized electrolyser (SDE), where the sulphur dioxide is oxidized at the anode to produce sulphuric acid and hydrogen ions (Equation [6]). The hydrogen ions pass through the membrane of the proton exchange membrane (PEM) electrolyser to the cathode, where they are reduced to form hydrogen gas (Equation [7]). The net electrolyser reaction therefore involves the conversion of sulphur dioxide and water into sulphuric acid and hydrogen (Equation [8]). The net HyS cycle reaction is the splitting of water into hydrogen and oxygen (Equation [9]), with the sulphuric acid cycling between the decomposition reactor and the electrolyser.
Thermal decomposition reaction at ± 900°C:
H2SO4(aq) → H2O(g) + SO2(g) + 1/2O2(g) [5]
Electro-oxidation of sulphur dioxide at the anode of the electrolyser:
S02(aq) +2H20(l) → H2SO4(aq) + 2H+ (aq)+2e [6]
Electro-reduction of hydrogen ions at the cathode of the electrolyser:
2H+(aq) + 2e → H2(g) [7]
Net electrochemical reaction at 80 - 120 °C (Equations [6] + [7]):
SO2 (aq) + 2H2O(l) → H2SO4 (aq) + H2(g) [8]
Net HyS cycle reaction (Equations 5 + 8):
H20(l) → H2(g) + 1/2O2(g) [9]
Between 1983 and 1999 the HyS process received no further interest due to the production and availability of cheap hydrogen through steam conversion from natural gas. The establishment of the Nuclear Hydrogen Initiative and Next Generation Nuclear Plant (NGNP) programmes in the USA, through the Office of Nuclear Energy, has however resulted in a renewed investigation into nuclear energy as a primary source of a future hydrogen economy. A renewed interest in the HyS process was sparked when the Office of Nuclear Energy of the US Department of Energy (DoE) identified 115 thermochemical hydrogen production cycles and rated the HyS cycle as the most promising (Summers and Gorensek, 2006).
From the schematic representation of the HyS process (Figure 3) it is clear that the feed includes water, heat, and electricity while the products consist of hydrogen and oxygen. There is, however, a strong drive to focus solely on the SDE, not only because this is where the most gains can be made, but because the SDE can serve as a SO2 sink and produce hydrogen (a clean energy carrier) in the process.
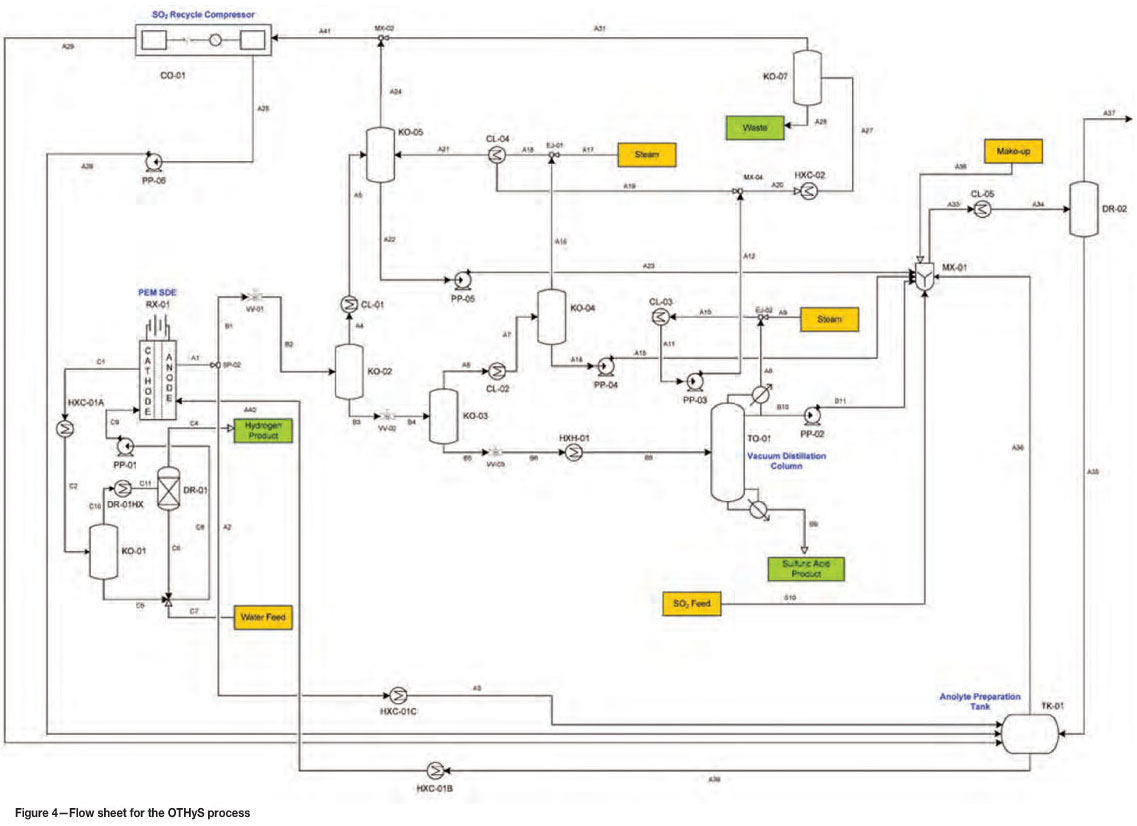
The Once-through Hybrid Sulfur (OTHyS) process
The OTHyS flow sheet (Figure 4) for only the SDE side of the HyS cycle was developed in Aspen assuming a coal-fired power station as a SO2 source. The specific power station values used were based on 6 χ 800 MWe supercritical coal-fired boilers, similar to Eskom's Medupi power station currently under construction in the Limpopo province in South Africa.
By eliminating the sulphuric acid decomposition step of the HyS cycle, the process is reduced in complexity due to fewer components and significant lower temperatures, with the highest process temperature being approximately 100°C in the SDE (note that the flow sheet has not been optimized).
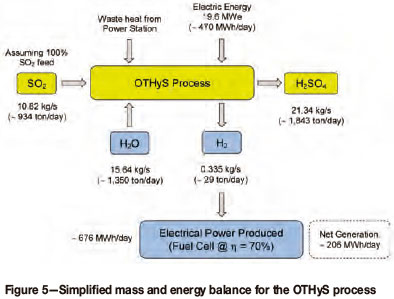
A high-level mass and energy balance was performed for the OTHyS flow sheet (Figure 4), and the results (Figure 5) show that for a SO2 feed of approximately 10.82 kg/s from the power station, hydrogen can be produced at a rate of approximately 0.335 kg/s. This hydrogen can be used, for example, to generate electricity via a fuel cell, and assuming a fuel cell efficiency of 70%, approximately 676 MWh of electricity can be produced per day. Given the electrical requirements of the OTHyS process for this application, which is in the order of 470 MWh/d, the net electricity generated is approximately 206 MWh/d. The economic feasibility of the process still needs to be verified, however.
The electrochemical oxidation of SO2
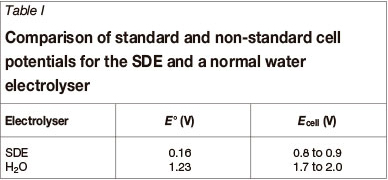
The electrochemical oxidation of SO2 (Equation [6]) has a standard reduction potential (Ε0) of 0.16V vs the standard hydrogen electrode (SHE). Standard conditions imply T= 298.15K (25°C), P = 1 bar, and an activity of 1 mol/l for each component. Under non-standard conditions the cell potential (Εcell) obviously changes, resulting in the electrolyser running at increased overpotentials (η). In contrast, regular water electrolysis has a theoretical reversible cell potential of 1.23 V (under standard conditions), which increases under non-standard conditions. A further 'constraint' that adds to the increase in overpotential (η) of water electrolysis is that electrolysis must be run at economically reasonable current densities (so as to increase the kinetics). It is for this reason that commercial water electrolysers operate at cell potentials of 1.7 to 2.0 V. Operating the SDE at reasonable current densities result in ohmic losses and electrode overpotentials (η), which causes the voltage to increase above 0.16 V. SDEs are therefore expected to run at cell potentials in the order of 0.8 to 0.9 V (Εcell). It is therefore quite clear that SO2 electrolysis operates at a potential of more than 1 V lower than normal water electrolysis. This is true for both standard and non-standard conditions (Table I). This means that the SDE consumes significantly less electricity to produce one mole of hydrogen compared to normal water electrolysis.
It was reported by Jeong and Kazimi (2007) that the SDE remains the main source of the HyS cycle inefficiency and that a 3% reduction of the cell potential will result in a 1% increase of overall cycle efficiency. Lu et al. (1981) predicted that cell potentials of 0.45-0.75 V could be obtained at current densities of 100-400 mA/cm2 for properly designed and optimized SDEs. Given their PEM SDE development experience, Savannah River National Laboratories (SRNL, 2007) reiterated the attainability of this target, stating that cell potentials of 0.6 V should be attainable if the SDE is run at higher operating temperatures (>100°C) and pressures (≥10 bar), still achieving economically reasonable current densities. According to SRNL their target is a cell potential of approximately 0.6 V at a current density of 500 mA/cm2 (Gorensek et al., 2009). These values, however, have been calculated taking the whole HyS cycle into account.
Notwithstanding the fact that no information could be obtained regarding catalyst loading studies, the current SDE design condition set by Gorensek et al. (2009) puts the current catalyst loading on the anode at 1 mg/cm2 Pt, with Pt being the current catalyst of choice.
Mechanism of SO2 electro-oxidation
In order to improve the effectiveness and economics of running a SDE, the operating cell potential, or overpotential, needs to be lowered while the current density, or electrode kinetics, needs to be increased. This is as a direct result of the slow rate of oxidation of SO2 at the anode, which contributes approximately 70% towards the overpotential. The largest source of improvement in SDE performance will arise from the identification of a means to increase electrode kinetics, i.e. the rate of oxidation of SO2 at the anode (resulting in increased current density). This is to be accomplished by the development of an improved electro-catalyst.
In identifying and/or developing such a novel electro-catalyst an in-depth understanding of the relevant chemical processes is crucial. Quijada et al. (2000) reported that numerous studies have been conducted to elucidate the mechanism that governs the oxidation of aqueous SO2 on platinum electrodes. Conflicting conclusions have, however, been reported on this topic in the early literature (Seo and Sawyer, 1965; Comtat and Mahenc, 1969; Appleby and Pichon, 1979; Audrey and Voinov, 1980), which can be attributed to the lack of adequate surface control and little attention paid to the influence of sulphur-containing adsorbates. Sulphur-containing adsorbed species are said to play a key role in improving the electrode kinetics of the oxidation of SO2 on platinum and gold (Spotnitz et al., 1983; Quijada et al., 1995; Samec and Weber, 1975a).
A review article published by O'Brien et al. (2010) on the electrochemical oxidation of aqueous SO2 with respect to the HyS cycle confirms and highlights the fact that there exists widespread disagreement in the literature on the mechanism of the electrochemical oxidation of aqueous SO2.
A possible reaction pathway may include the following reaction steps, with M representing an active site on the electrode surface (Appleby and Pinchon, 1979; Lu and Ammon, 1982):
Μ + H2O → MOH + H+ + e- [10]
MOH →ΜΟads + Η+ + e- [11]
SO2+ H2O + MOads → H2SO4 + Μ [12]
It is widely accepted that the electrochemical oxidation of aqueous SO2 proceeds according to the following reaction pathway (Equation [6]):
SO2(aq) + 2H2O(l) → H2SO4(aq) + 2H+(q) + 2e- [13]
In a slight modification, Samec and Weber (1975b) suggest a potential oxidation pathway of aqueous SO2 to firstly form HSO4-, which then combines with one proton to form sulphuric acid; the net result being that of Equation [6]. In another variation, Seo and Sawyer (1965) suggest that the oxidation pathway is probably the oxidation of SO2 to SO42-, which in turn combines with two protons sequentially to again form sulphuric acid; the net result again being that of Equation [6]. Although all three of the above studies found aqueous SO2 to oxidize according to the same net chemical equation, the reaction pathways proceed via differing intermediate compounds. It is unknown what the lifetimes of these intermediates are, i.e. which reaction determines the overall rate of the net equation, which in turn could have a negative impact due to possible side reactions.
Valensi et al. (1966 and 1973) studied the thermodynamics of the sulphur-water system and classified different sulphur compounds as being stable or metastable. According to the authors, only sulphur (S), sulphuric acid (H2SO4), and hydrogen sulphide (H2S) are stable. Compounds classed as metastable can undergo further oxidation or reduction, depending on what is thermodynamically favourable. The following compounds, in acid solution, are all metastable: S2O32-, S2O42-, S2O62-, and S4O62-. According to Valensi, only the dithionate ion (S2O62-) could be produced by oxidation of SO2, with the dithionate ion in turn being reduced to form H2SO3. The net result is then the oxidation of aqueous SO2 to produce H2SO3 (Equations [14] -[16]).
2S02+2H2O → S2O62-+4H* + 2e- [14]
S2062- + 4H+ + 2e → 2H2SO3 [15]
SO2 +H2O → H2SO3 [16]
This pathway is, however, highly unlikely as H2SO3 can be formed only under cryogenic conditions, is highly unstable at room temperature and above, and to that regard has never been isolated (Voegele et al., 2004). All of these compounds, except tetrathionate (S4O62-), can however be prepared electrochemically (Zhdanov, 1975). It is therefore clear that a number of unwanted compounds can be produced that (a) could poison both the catalyst and the membrane, in other words the MEA (membrane electrode assembly), and (b) result in a lowering of the current density as these 'side reactions' consume charge. Of these unwanted products both sulphur (S) and hydrogen sulphide (H2S) pose the biggest threat. Sulphur deposition could proceed according to the following reaction pathway, with sulphurous acid (H2SO3) being an unstable intermediate (Equations [17]-[19]).
SO2 + H2O → H2SO3 [17]
H2SO3 + 4H+ + 4e- → S + 3H2O [18]
SO2 + 4H* + 4e- → S + 2H2O [19]
The above-mentioned deposition of sulphur clearly follows an electrochemical pathway. This thermodynamic instability of SO2 has been highlighted by Noyes and Steinour (1929) with a non-electrochemical deposition of sulphur occurring according to the following disproportionation reaction (Equation [20]).
3SO2 + 2H2O → S + 2H2SO4 [20]
This reaction does, however, proceed at a negligible rate except at elevated temperatures, i.e. over 100°C, which is within the operating temperature of the SDE. The Pourbaix diagram of the sulphur-water system at 100°C and 6 bar pressure clearly indicates that both S and H2S are stable within the pH and potential operating range of the SDE (Figure 6).
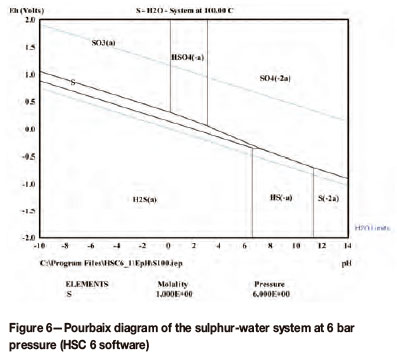
Adsorbed sulphur species play a key role in both the electrochemical oxidation (at the anode) and the electrochemical reduction (at the cathode) of SO2, which impacts the electrode kinetics of these processes. An improved understanding of these processes will therefore benefit both the anodic oxidation of SO2, as well as the cathodic reduction of SO2 with the subsequent poisoning of the cathode, which impacts negatively on the reduction of hydrogen ions to form hydrogen gas.
It has been shown that the rates of some electrocatalytic (Loucka, 1972) and redox (Samec and Weber, 1972 and 1973) reactions are considerably changed by the presence of adsorbed sulphur on the surface of the platinum catalyst. Spotnitz et al. (1983) conducted a study of SO2 oxidation at a Pt rotating disc electrode (RDE) in sulphuric acid solutions of 0.5-6.0 M at ambient temperatures so as to characterize the effect of surface pretreatments. The authors state that the SO2 oxidation reaction has been the focus of many investigations, but the results are not in quantitative agreement and the mechanism of the reaction remains in doubt. There are many factors that complicate the voltammetric behavior of this system, e.g. (i) it is possible that SO2 reduction may occur in the same potential region in which oxidation occurs, (ii) surface oxide formation at more positive potentials, (iii) the requirement for pretreatment and activation of the electrode/catalyst, and (iv) the thermodynamics of the sulphur-water system. The authors provide evidence that it is the presence of reduced sulphur dioxide species on the electrode surface that determines the course of the anodic oxidation.
Experimental electrocatalyst research and development
The research and development of the electrocatalyst for the electro-oxidation of sulphur dioxide has until now involved only single-electrode studies. These typically involve RDE studies in a three-electrode electrochemical cell probing a specific catalyst's activity by means of linear polarization and cyclic voltammetry. The generous funding of Anglo American Platinum, the North-West University, and the Department of Science and Technology, through the HySA Infrastructure Center of Competence, has allowed the PGM Group at the Potchefstroom campus of the North-West University to acquire equipment and set up a unique and dedicated laboratory to synthesise and probe the electrocatalytic activity of up to 64 different electrocatalysts simultaneously and under the same conditions (see Combinatorial study).
Single-electrode studies
With regard to the catalyst required for the electrochemical oxidation of aqueous SO2, O'Brien et al. (2010) state that there has been relatively little catalyst development. Catalysts investigated thus far include carbon (C), platinum (Pt), platinum-aluminum (Pt/Al), palladium (Pd), and gold (Au). Some initial results, obtained by the PGM Group employing polycrystalline platinum, palladium, and rhodium (Rh) are reported here.
Seo and Sawyer (1965) report that successful electrochemical oxidation of SO2 in acidic media with a platinum electrode requires an electrode preconditioning procedure, which involves scanning three or four times between -0.15 V and +1.5 V (SCE) directly in the sample solution producing the required active electrode surface. This 'activation' allows (i) for the current/voltage traces to be reproducible, (ii) for the current/voltage traces to have reached their maximum with regard to the limiting peak current, (iii) that the slope Δί/ΔV has reached a maximum, and (iv) that the overvoltage has reached a minimum. For platinum, the initial scan to high anodic voltage causes an oxide film to be formed on the electrode surface. When the repeat scan is started at a negative voltage, corresponding to that at which hydrogen evolution begins, the oxide is again stripped. It is this stripped electrode that has the desired surface characteristics. The electrochemical behaviour of a gold anode in acidic solution is also dependent on its surface state. However, because of its less noble character its surface is largely destroyed by repeated anodic polarization cycles, especially in a chloride medium. Although anodic activation is still required for the successful oxidation of aqueous SO2, careful stripping of the electrode surface before the following scan is not necessary, as gold surface oxide films dissolve easily in acidic solution. Care should, however, be taken to avoid extensive cathodization, which removes the desired activity of the electrode surface.
According to Samec and Weber (1975b), both the reproducibility and magnitude of the oxidation current are strongly dependent on whether the electrode is cyclically prepolarized directly in the SO2 solution. After such electrode pretreatment, changes in the rate of electrochemical reactions may occur due to factors modifying the properties of the metal/solution interface, such as (a) changes in the active surface area of the electrode, and (b) changes in the degree of adsorption of particles on the surface from (i) the base electrolyte (e.g. oxygen, base electrolyte ions, impurities) and (ii) electrochemical reactions of the studied substance.
Studies conducted by Appleby and Pinchon (1980) and Lu and Ammon (1980) have found carbon materials (graphite and carbides) to be catalytically inactive with regard to the electrochemical oxidation of aqueous SO2. Activated carbon and carbon black have, however, been found to have intermediate activity (Appleby and Pinchon, 1980). Wiesener (1973) found that the treatment of carbon at high temperatures and under various oxidizing gas streams resulted in improved catalytic activity in itself.
Investigating the noble metals as catalysts for the electrochemical oxidation of aqueous SO2, Appleby and Pinchon (1980) found platinum to have higher activity than palladium. This was confirmed by Lu and Ammon (1980), who furthermore suggest that the formation of PdO on the electrode is advantageous for the reaction. A study conducted by Colon-Mercado and Hobbs (2007) found that Pt/C exhibited lower activation energies with higher current densities than Pd/C, which furthermore proves the enhanced catalytic activity of Pt above that of Pd. Pd, furthermore, seems to be less stable than Pt, as the authors noticed some dissolution of the Pd electrode.
Contrary to the previous findings, Lu and Ammon (1982) reported that a PdOx/C electrode exhibited higher limiting current density and lower polarization potentials compared to a Pt-black/C electrode. It has to be noted that Pt-black is generally regarded as a superior catalyst to PdO, which makes their observation unexpected and interesting. They mention, for example, that at 1 mA/cm-2 the anodic overpo-tential on the PdOx/C electrode is approximately 50 mV less than on the Pt-black/C electrode.
A study conducted by Lee and Langer (1995) found that the doping of platinum with a small amount of aluminum (Al) resulted in a large improvement in electrode performance. The catalyst is believed to be bimetallic and not alloyed. An unexpected observation was made by Quijada and Vazquez (2000) when they found that gold (Au) was inherently more active than platinum (Pt) for the electrochemical oxidation of aqueous SO2. The authors ascribe this to a difference in the oxidation mechanism. Samec and Weber (1975b) studied the electro-reduction of SO2 on gold for the first time. They found that SO2 reduces at E < 0.4 V (RHE), which gives rise to two consecutive cathodic peaks. The authors associated the more positive potential peak with the adsorption of sulphur with a maximum coverage of 0.25 (1 representing total coverage). The less positive potential peak was associated with a six-electron transfer resulting in the generation of H2S, which adsorbs with a maximum coverage of 0.45. Both these species, i.e. elemental sulphur and H2S, coexist on the surface in a wide potential region. The electrochemical reduction of SO2 on gold was also investigated by Wilke et al. (1991) to identify the chemical nature of adsorbed intermediates. They identified a metal-sulphur (MS) stretch mode as well as sulphur-sulphur (S-S) stretch and bend modes for polysulphides.
Preconditioning of both platinum and gold electrodes has an effect on the catalytic activity of the electrode. This is portrayed in Figure 7 (for gold) and Figure 8 (for platinum). With regard to the current response, two regions of oxidation have been specified, defined by their potential intervals, and have been labeled I and II. Region I has been assigned to lower potentials and coincides with a double layer region, whereas region II has been assigned to higher potentials and coincides with the potential range of surface oxide formation. These graphs also indicate three different polarization scenarios with each scenario defined by its lower potential limit (Elow). These three scenarios can qualitatively be defined as follows:
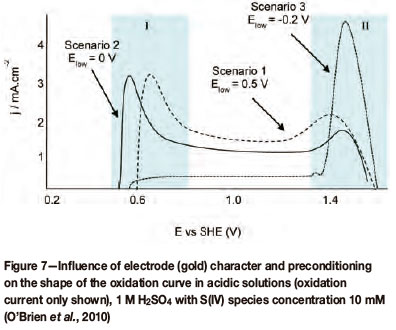
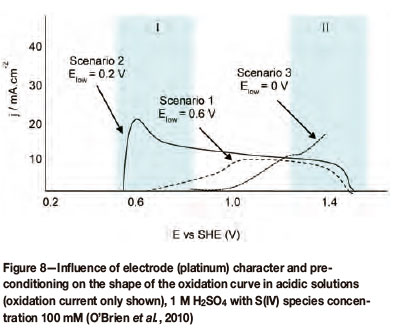
Scenario 1: limited response in regions I and II (high Elow value)
Scenario 2: enhanced/activated response in region I (mid-range Elow value)
Scenario 3: inhibition of response in region I, changed response in region II (low Elow value).
Elow is defined as the lowest applied potential the electrode was cycled to, and in some cases it is the potential at which the electrode was held for a specified time prior to cycling of the electrode. From Figure 5 (for gold) it is clear that the catalytic region is defined by regions I and II, which coincides with preconditioning scenarios 2 and 3.
From Figure 8 (for platinum) the same is evident in that the catalytic region is defined by regions I and II, which coincide with preconditioning scenarios 2 and 3. It is clear that catalytic activity, indicated by a high or higher current density, depends on the preconditioning of the electrode by means of potential cycling or potential holding for a specific time prior to cycling.
Oxidation of SO2 at high potentials (region II) has also been attributed to the 'chemical' oxidation of SO2 by adsorbed platinum oxide (Matveeva and Kasatkin, 1984; Korzeniewski and McKenna, 1987) according to the following reaction (Equation [21]).
Pt (O)ads + S02+ 3H20 → H2S04+ 2H2O + Pt [21]
Quijada et al. (1995), however, opposed the above and proposed a mechanism whereby SO2 and H2O compete for active Pt sites, and it is the occupation of these active sites by oxygen-containing species that causes the inhibition.
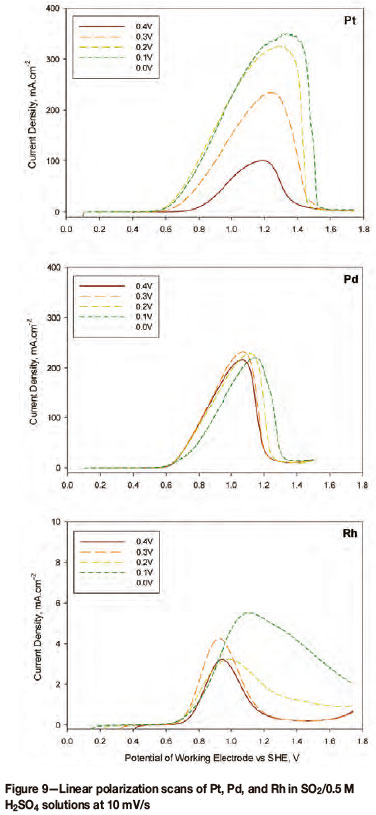
Comparing the electro-oxidation of SO2 on platinum, palladium, and rhodium, it has been found that the starting potential of the forward linear polarization scan affects the peak current density obtained (Figure 9). As the starting potentials are below 0.45 V, sulphur is deposited onto the catalyst through the reduction of sulphur dioxide. The different starting potentials cause different amounts of sulphur to be deposited and thereby affect the forward scan differently. In all instances SO2 was bubbled for 30 minutes into a 0.5 M H2SO4 electrolyte solution, and the linear polarization scans were conducted at 10 mV/s (all electrodes being 5 mm in diameter).
It has been conclusively shown that catalytic oxidation of SO2 (scenario 2) is a result of sulphur formation (Figure 10). There is therefore a high dependence of the oxidation reaction on Elow, which determines the degree of sulphur coverage. Excessive sulphur formation (layers exceeding a bilayer) and the possible formation of polysulphides at lower potentials (scenario 3) inhibit the reaction.
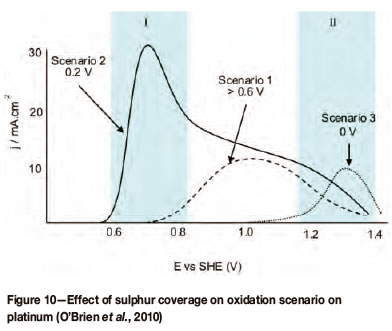
To prevent this electrochemical deposition of sulphur (Equation [22]), the potential has to be kept below the calculated potential of 0.449 V (according to the Gibbs equation). The literature would, however, seem to be in disagreement as to the exact requirement.
SO2 + 4H* + 4e- → S + 2H2O [22]
Wilke et al. (1991) suggest keeping the potential above 0.15 V measured against the standard hydrogen electrode (SHE) as reference. Loucka (1971) in turn suggests keeping the potential above 0.375 V (SHE), whereas Quijada and Vazquez (2000) found that potentials below the open circuit potential (OCP) of 0.65 V in 2 M H2SO4 will result in reduction currents, i.e. the deposition of sulphur at the cathode. The OCP is, however, determined by various factors including H2SO4 concentration, the electrolyte, temperature, cell resistance, etc., and will therefore vary depending on the prevailing conditions.
Combinatorial strategy/plan
As mentioned earlier, a unique and dedicated laboratory has been set up at the North-West University for the research and development of electrocatalysts for the electro-oxidation of sulphur dioxide. Some of the key components are depicted in Figure 11.


A combinatorial sputter deposition system (Figure 11a), from PVD Products, is a unique tool with which to explore new chemistries in thin film form. Using multiple sputter sources, each loaded with a different material, binary, tertiary, and quaternary material systems can be easily explored by computer control. The heart of the system is a programmable X-Y stage that holds wafers up to 4 inches in diameter. A metal mask with an aperture of 4 x 4 mm square sits directly in front of the wafer. Four 1 inch diameter magnetrons, each loaded with metallic or oxide materials (or other materials), are directed at the aperture. Using software to set the power levels (either RF or DC) to any selected magnetron, the chemical composition of the deposited 4 x 4 mm square film can be varied over a wide range. Using a step and repeat process on the X-Y stage the wafer can be repositioned to a new location and a new chemistry can be deposited. The compositional changes should be as small as 1% or less, depending on material properties and deposition conditions.
The system can be programmed to provide an 8 x 8 array of 4 mm square samples, each with a different composition. Thus, 64 different chemistries (Figure 11b) can be deposited in one pump-down cycle of the system. Depositing films on substrates that include electrodes, each of the 64 chemical compositions can be evaluated simultaneously under the same conditions within an electrochemical cell (Figure 11c) for the electrocatalytic oxidation of sulphur dioxide. The electrocatalysts that will be investigated will be platinum group metal (PGM) based, i.e. combinations of the platinum group metals (noble metals) with other transition metals. Each of the 64 chemical compositions (electrocatalysts) will be linked to a dedicated potentiostat, as part of a 64 channel potentiostat (Figure 11d), which will allow the simultaneous electrochemical testing/screening of these PGM based electro-catalysts. Coupled with this set-up one can quickly sample a wide range of chemistries and find those that give the desired electrical properties, i.e. the highest current density at the lowest input potential. This can be depicted as hotspots (Figure 12) similar to that of the electro-oxidation of methanol as conducted by Cooper and McGinn (2006). Such a system will greatly reduce the time it takes to find the desired chemical compound with the required electrical (or other) properties. The first results from this system are expected in 2013.
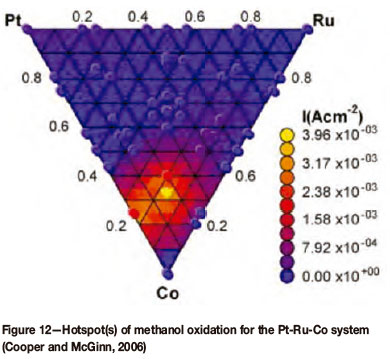
This equipment and laboratory is, however, not specific to a single electrochemical reaction, e.g. the electro-oxidation of sulphur dioxide, but could be well applied for the research and development of electrocatalysts for other important reactions. These include the electrocatalytic reduction of oxygen, a crucial step in the electrochemical production of electrical energy in fuel cells.
Theoretical electrocatalyst research and development
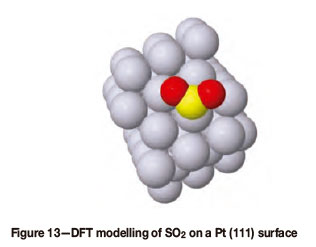
The number of different chemical combinations that can be considered and investigated by means of a combinatorial electrocatalyst synthesis and screening set-up is quite substantial. To identify a superior electrocatalyst for any electrochemical reaction is really like looking for the proverbial needle in a haystack. There are just so many combinations, and hitting on the right one experimentally could come down to pure luck in many instances. This is where theoretical modelling comes into play, which has shown to be quite successful in identifying electrocatalysts with superior catalytic activity for the oxygen reduction reaction (Nørskov et al., 2004). Modelling has been conducted by the PGM Group of the North-West University and the CAMD group of the Technical University of Denmark, employing density functional theory (DFT) to calculate the binding energies of different intermediates, i.e. OH, S, SO2, and HSO3, to the surface of pure metal slabs (Figure 13).
The following reaction mechanism was considered whereby (a) the reduction of SO2 to S has to be prevented (Equation [23]), (b) SO2 has to be converted into HSO3 through the splitting of one water molecule (Equation [24]), and (c) HSO3 is converted to H2SO4 through the splitting of a second water molecule (Equation [25]).
SO2 + 4H* + 4e- → S + 2H2O [23]
SO2 + H2O → HSO3+ H* + e- [24]
HSO3+ H2O → H2S04+ H* + e- [25]
Preliminary results would indicate that the binding energies of S, SO2, and HSO3 all scale with the binding energy of OH on the different metal surfaces (Figure 14). It is, however, clear that for both silver (Ag) and gold (Au) the different components do not bind as strongly and are therefore not following the trend.
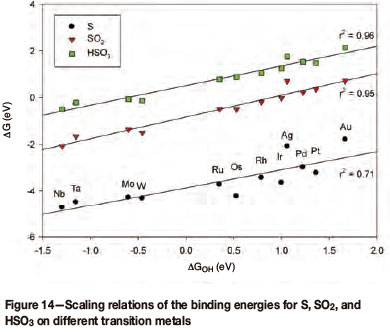
If both Ag and Au are removed from the scaling relations for S, SO2, and HSO3 the linearities for all three lines improve to r2 = 0.98, r2 = 0.98, and r2 = 0.84 respectively. Converting these binding energies to potential and plotting against the OH-binding energy yields a volcano plot, based on the abovementioned preliminary results (Figure 15).
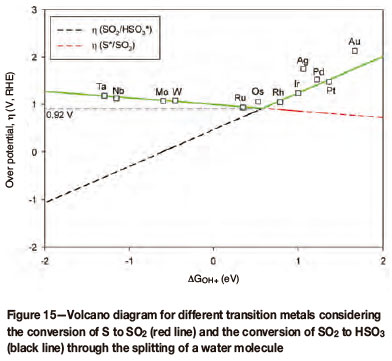
Considering pure transition metals, this would indicate that osmium (Os) should thermodynamically have the highest activity for the electro-oxidation of sulphur dioxide, as it is at this point that the lowest potential of 0.92 V is obtained for preventing the reduction of SO2 to S and, at the same time, ensuring the splitting of a water molecule resulting in the formation of HSO3. The second water molecule is split more easily than the first, and in that regard the thermodynamics is downhill subsequent to the splitting of the first water molecule. This theoretical prediction is about to be experimentally tested, and DFT modelling of the electro-oxidation of sulphur dioxide on other novel metal surfaces is in progress.
Conclusions
The electro-oxidation of sulphur dioxide holds the potential for converting sulphur dioxide into sulphuric acid and hydrogen gas. This is in contrast to the regular abatement technologies, whereby sulphur dioxide is converted into either sulphuric acid or a salt. Electro-oxidation holds the promise of a potential 'return on investment' in that the hydrogen gas is a clean energy carrier that could be used in, for example, a fuel cell for backup power. The biggest contribution to be made in developing this technology is the identification of a suitable electrocatalyst to increase the current density and at the same time reducing the overpotential. Investigations thus far reveal that the preconditioning of the electrocatalyst is crucial, and in that regard sulphur deposition as well as the adsorption of sulphur-based species has a key role to play in the reaction mechanism and in understanding and improving the activity of the electro-catalyst. To date very little work has been done on the research and development of an effective electrocatalyst and work is in progress conducting experimental studies, i.e. single-electrode studies and combinatorial electrocatalyst synthesis and screening studies, as well theoretical studies employing density functional theory. First indications, according to density functional theory, are that an electro-catalyst other than platinum might be identified that could improve not only the catalytic activity, but also the economics of this technology.
Acknowledgements
The generous funding from Anglo American Platinum, the North-West University, and the Department of Science and Technology, through the HySA Infrastructure Center of Competence, is gratefully acknowledged.
References
Appleby, A.J. and Pinchon, B. 1979. The mechanism of the electrochemical oxidation of sulfur dioxide in sulfuric acid solutions. Journal of Electroanalytical Chemistry, vol. 95. pp. 59-71. [ Links ]
Appleby, A.J. and Pinchon, B. 1980. Electrochemical aspects of the H2SO4 SO2 thermoelectrochemical cycle for hydrogen production. International Journal of Hydrogen Energy, vol. 5. pp. 253-267. [ Links ]
Audrey, C. and Voinov, M. 1980. Inhibitions of the SO2 electrochemical oxidation reactions on platinum in sulfuric acid solutions. Electrochimica Acta, vol. 25. pp. 299-301. [ Links ]
Bezuidenhout, G.A., Davis, J., van Beek, B., and Eksteen, J.J. 2012. Operation of a concentrated mode dual-alkali scrubber plant at the Lonmin smelter. Journal of the Southern African Institute of Mining and Metallurgy, vol. 112. pp. 657-665. [ Links ]
Brecher, L.E. and Wu, C.K. 1975. Electrolytic decomposition of water, Westinghouse Electric Corporation. US Patent No. 3888750. [ Links ]
Bunce, N. 1994. Environmental Chemistry. 2nd edn. Wuerz Publishing. p. 376. [ Links ]
Calvert, J.G. and Stockwell W.R. 1984. Mechanisms and rates of gas-phase oxidations of sulfur dioxide and nitrogen oxides in the atmosphere. SO2, NO and NOx Oxidation Mechanisms: Atmospheric Considerations. Calvert, J.G. (ed.). Butterworths. pp. 1-62. [ Links ]
Colon-Mercado, H.R. and Hobbs, D.T. 2007. Catalyst evaluation for a sulfur dioxide-depolarized electrolyzer. Electrochemistry Communications, vol. 9. pp. 2649-2653. [ Links ]
Comtat, M. and Mahenc, J. 1969. Electrochemical Oxidation of Sulfur Dioxide on a Platinum Electrode. Bulletin de la Société Chimique de France, vol. 11. p. 3862. [ Links ]
European Commission, Joint Research Centre (JRC)/Netherlands Environmental Assessment Agency (PBL). 2010. Emission Database for Global Atmospheric Research (EDGAR). Release version 4.1. http://edgar.jrc.ec.europa.eu [ Links ]
Fang, Y., Zeng, Y., and Li, S. 2008. Technological influences and abatement strategies for industrial sulphur dioxide in China. International Journal of Sustainable Development & World Ecology, vol. 15, no. 2. pp. 122-131. [ Links ]
Friend, J.P. 1973. The global sulfur cycle. Chemistry of the Lower Troposphere. Rasool, S.I. (ed.). Plenum Press, New York, NY. pp. 177-201. [ Links ]
Gorensek, M.B., Summers, W.A., Bolthrunis, C.O., Lahoda, E.J., Allen, D.T., and Greyvenstein, R. 2009 Hybrid Sulfur Process Reference Design and Cost Analysis - Final Report, SRNL-L1200-2008-00002. Rev 1, 12 June. [ Links ]
Jeong, Y.H. and Kazimi, M.S. 2007. Optimization of the Hybrid Sulfur Cycle for nuclear hydrogen generation. Nuclear Technology, vol. 159. pp. 147-157. [ Links ]
KORZENIEWSKI, C., McKenna, W., and Pons, S. 1987. An in situ infrared study of the oxidation of sulfur dioxide on platinum electrodes. Journal of Electroanalytical Chemistry and Interfactial Electrochemistry, vol. 235. pp. 361-368. [ Links ]
Kruger, B. 2004. Recovery of SO2 from low strength off-gases. International Platinum Conference 'Platinum Adding Value'. Southern African Institute of Mining and Metallurgy, Johannesburg. pp. 59-62. [ Links ]
Lee, J. and Langer, S.H. 1995. Electrochemical sulphur dioxide oxidation with platinum-aluminum electrocatalysts. Journal of Applied Electrochemistry, vol. 25. pp. 353-357. [ Links ]
Loucka, T. 1971. Adsorption and oxidation of sulphur and of sulphur dioxide at the platinum electrode. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry, vol. 31. pp. 319-332. [ Links ]
Loucka, T. 1972. Adsorption and oxidation of organic compounds on a platinum electrode partly covered by adsorbed sulphur. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry, vol. 36. pp. 355-367. [ Links ]
Lu, P.W.T. and Ammon, R.L. 1980. An investigation of electrode materials for the anodic oxidation of sulfur dioxide in concentrated sulfuric acid. Journal of the Electrochemical Society, vol. 127. pp. 2610-2616. [ Links ]
Lu, P.W.T., Garcia, E.R., and Ammon, R.L. 1981. Recent developments in the technology of sulfur dioxide depolarised electrolysis. Journal of Applied Electrochemistry, vol. 11. pp. 347-355. [ Links ]
Lu, P.W.T. and Ammon, R.L. 1982. Sulfur dioxide depolarized electrolysis for hydrogen production: development status. International Journal of Hydrogen Energy, vol. 7, no. 7. pp. 563-575. [ Links ]
Matveeva, E.S. and Kasatkin, E.V. 1984. Adsorption of sulfur dioxide on the platinum electrode and influence on its electrooxidation in concentrated solutions. Soviet Electrochemistry, vol. 19, pp. 801-805. [ Links ]
Noyes, A.A. and Steinour, H.H.1929. The potential of inert electrodes in solutions of sulfurous acid and its behavior as an oxidizing and reducing agent. Journal of the American Chemical Society, vol. 51. pp. 1409-1428. [ Links ]
O'Brien, J.A., Hinkley, J.T., Donne, S.W., and Lindquist, S-E. 2010. The electrochemical oxidation of aqueous sulfur dioxide: A critical review of work with respect to the hybrid sulfur cycle. Electrochimica Acta, vol. 55. pp. 573-591. [ Links ]
World Bank. 1999. Pollution Prevention and Abatement Handbook. Geneva. [ Links ]
Quijada, C., Rodes, A., Vazquez, J.L., Perez, J.M., and Aldaz, A. 1995. Electrochemical behaviour of aqueous SO2 at Pt electrodes in acidic medium. A voltammetric and in situ Fourier transform IR study Part I. Oxidation of SO2 at Pt electrodes with sulphur-oxygen adsorbed species. Journal of Electroanalytical Chemistry, vol. 394. pp. 217-227. [ Links ]
Quijada, C., Morallon, E., Vazquez, J.L., and Berlouis, L.E.A. 2000. Electrochemical behaviour of aqueous SO2 at polycrystalline gold electrodes in acidic media. A voltammetric and in-situ vibrational study. Part II. Oxidation of SO2 on bare and sulphur-modified electrodes. Electrochimica Acta, vol. 46. pp. 651-659. [ Links ]
Quijada, C. and Vazquez, J.L. 2000. Recent Research Developments in Electrochemistry, Pandalai S.G., vol. 3. p. 137. [ Links ]
Samec, Z. and Weber, J. 1972. Reduction of ferric ion on a rotating platinum electrode of the turbulent type in the presence and absence of adsorbed sulphur. Journal of Electroanalytical Chemistry, vol. 38. pp. 115-126. [ Links ]
Samec, Z. and Weber, J. 1973. The influence of chemisorbed sulphur on the kinetic parameters of the reduction of Fe3+ ions on a platinum electrode on the basis of the marcus theory of electron transfer. Journal of Electroanalytical Chemistry, vol. 44. pp. 229-238. [ Links ]
Samec, Z. and Weber, J. 1975a. Study of the oxidation of SO2 dissolved in 0.5 M H2SO4 on a gold electrode - II rotating disc electrode. Electrochimica Acta, vol. 20. pp. 413-419. [ Links ]
Samec, Z. and Weber, J. 1975b. Study of the oxidation of SO2 dissolved in 0.5 M H2SO4 on a gold electrode - I stationary electrode. Electrochimica Acta, vol. 20. pp. 403-412. [ Links ]
Seo, E.T. and Sawyer, D.T. 1965. Electrochemical oxidation of dissolved sulphur dioxide at platinum and gold electrodes. Electrochimica Acta, vol. 10. pp. 239-252. [ Links ]
Sivasubramanian, P., Ramasamy, R.P., Freire, F.J., Holland, C.E., and Weidner, J.W. 2007. Electrochemical hydrogen production from thermochemical cycles using a proton exchange membrane electrolyzer. International Journal of Hydrogen Energy, vol. 32. pp. 463-468. [ Links ]
Smith, S.J., van Aardenne, J., Klimont, Z., Andres, R.J., Volke, A. and Delgado Arias, S. 2011. Anthropogenic sulfur doxide emissions: 1850-2005. Atmospheric Chemistry and Physics, vol. 11. pp. 1101-1116. [ Links ]
Spotnitz, R.M., Colucci, J.A., and Langer, S.H. 1983. The activated electro-oxidation of sulfur dioxide on smooth platinum. Electrochimica Acta, vol. 28, no. 8. pp. 1053-102. [ Links ]
Savannah River National Laboratory (SRNL). 2007. SRNL achieves milestone en route to hydrogen economy. Jackson, SC. [ Links ]
Srivastava, R.K., Jozewicz, W, and Singer, C. 2001. SO2 Scrubbing Technologies: A Review. Environmental Progress, vol. 20, no. 4. pp. 219-228. [ Links ]
Su, S., Li, B., Cui, S., and Tao, S. 2011. Sulfur Dioxide Emissions from Combustion in China: From 1990 tp 2007. Environmental Science and Technology, vol. 45. pp. 8403-8410. [ Links ]
SuMMERS, W.A. and GORENSEK, M.B. 2006. Nuclear hydrogen production based on the Hybrid Sulfur Thermochemical Process. Proceedings of ICAPP '06, Reno, Nevada, 4-8 June. Paper 6107. [ Links ]
Valensi, G., Van Muylder, J., and Pourbaix, M. 1966. Atlas of Electrochemical Equilibria in Aqueous Solutions. Pourbai, M. (ed.). Vol II. Pergamon Press, Oxford. p. 545. [ Links ]
Valensi, G. 1973. Technical Report RT 207 for Centre Belge d'Etude de la Corrosion. p. 121. [ Links ]
Voegele, A.F., Loerting, T., Tautermann, C.S., Hallbrucker, A., Mayer, E., and Liedl, K.R. 2004. Sulfurous acid (H2SO3) on Io? Icarus, vol. 169. pp. 242-249. [ Links ]
WBK & Associates Inc. 2003 Sulphur Dioxide: Environmental Effects, Fate and Behaviour. Alberta Environment, Government of Alberta, Edmonton, AB. [ Links ]
Westcott, G., Tacke, M., Schoeman, N., and Morgan, N. 2007. Impala Platinum Smelter, Rusternburg - an integrated smelter off-gas treatment solution. Journal of the Southern African Institute of Mining and Metallurgy, vol. 107. pp. 281-287. [ Links ]
Wiesener, K. 1973. The electrochemical oxidation of sulphur dioxide at porous catalysed carbon electrodes in sulphuric acid. Electrochimica Acta, vol. 18. pp. 185-189. [ Links ]
Wilke, T., Gao, X., Takoudis, C.G., and Weaver, M.J. 1991. Surface-enhanced Raman spectroscopy at transition metal-gas interfaces: adsorption and reactions of sulfur dioxide on platinum-, rhodium-, and ruthenium-coated gold. Journal of Catalysis, vol. 130. pp. 62-75. [ Links ]
Zhdanov, S.I. 1975. Encyclopedia of the Electrochemistry of the Elements. Bard, A.J. (ed.). Marcel Dekker, New York. p. 273. [ Links ]
© The Southern African Institute of Mining and Metallurgy, 2013. ISSN 2225-6253.
This paper was first presented at the, 4th Sulphur & Sulphuric Acid 2013 Conference, 3-4 April 2013, Sun City, Pilanesberg, South Africa.


{kind=link}
{kind=link}