










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkJournal of the Southern African Institute of Mining and Metallurgy
On-line version ISSN 2411-9717
Print version ISSN 2225-6253
J. S. Afr. Inst. Min. Metall. vol.112 spe Johannesburg Jul. 2012
JOURNAL PAPER
Platinum promotion of AU/AI₂O₃ catalysts for glycerol oxidation: activity, selectivity and deactivation
M. Royker; J. Case; E. van Steen
Centrefor Catalysis Research, Department of Chemical Engineering, University of Cape Town
SYNOPSIS
Gold has been demonstrated as a possible catalyst for oxidation reactions. Some evidence for a possible promotion effect of platinum has also been recorded. The influence of platinum as promoter for Au/y-Al2O3 prepared via anionic ion-exchange for the oxidation of glycerol was investigated in a batch reactor at 60°C. It is inferred that the addition of platinum reduces the catalytic activity and the rate of deactivation, resulting in an overall higher final conversion of glycerol with increasing platinum loading. The addition of platinum to the catalyst favours the formation of the desired product, glyceric acid.
Keywords: gold-platinum catalysts, bimetallic catalysts, alumina, glycerol, oxidation.
Introduction
The production of biodiesel via trans-esterification of triglycerides yields glycerol as a side-product (Ma and Hanna, 1999). Approximately 100 kg of glycerol is formed per ton of biodiesel produced. Hence, the commercial viability of biodiesel production also depends on the available market for glycerol. Although glycerol itself is a useful chemical used in drugs, oral care products and food, (Morrison, 2000), its market is far too small to take up a significant amount of glycerol produced from a growing biodiesel market, necessitating the development of glycerol as a platform chemical (Zhou et al., 2008). Selective oxidation of glycerol (preferably using air) is the preferred method to generate other chemical building blocks, such as dihydroxyacetone, glyceric acid, and tartronic acid.
Supported gold catalysts have been shown to be active for the selective oxidation of glycerol to glyceric acid using oxygen (Carrettin et al., 2003; Porta and Prati, 2004; Demirel-Gülen et al., 2005; Demirel et al. 2007a). The oxidation of alcohols and polyols over gold catalysts requires alkaline conditions (Carretin et al., 2003), since the first step in the oxidation is the proton abstraction from glycerol (Ketchie et al., 2007). Furthermore, oxygen from the hydroxyl ion is incorporated into the product acid. The hydroxyl ion is regenerated via O2 reduction with water (Zope et al., 2010).
The catalytic oxidation of glycerol can yield a multitude of products via a series of consecutive and parallel reactions. Control of product selectivity is of great importance to minimize the required separation processes to obtain the pure product compounds. Glycerol can be oxidized to glyceraldehyde, which is under basic conditions rapidly converted into dihydroxyacetone. Glyceric acid can be formed either directly from glycerol or in a consecutive reaction involving glyceraldehyde. The consecutive oxidation of glyceric acid yields tartronic acid.
The oxidation of glycerol may lead to the formation of degradation products such as glycolic acid, oxalic acid, acetic acid, formic acid, and COx. Supported gold catalysts have been reported to be selective towards the formation of glyceric and glycolic acid (Porta and Prati, 2004; Ketchie et al., 2007), with the latter formed directly from glycerol in a parallel pathway.
Bimetallic gold-platinum catalysts have been reported to be more active than monometallic gold-based catalysts (Bianchi et al., 2005; Demirel et al., 2007a) increasing the activity (expressed per gram of metal) by up to 80 percent. A similar enhancement was observed by alloying gold with palladium (Dimitratos et al., 2006b).
Supported gold catalysts are typically supported on carbon. However, the harsh conditions experienced typically in liquid-phase reactions require a highly mechanically stable support (Villa et al., 2010). In this study, the performance of monometallic gold and bimetallic Au-Pt catalysts supported on alumina for the liquid-phase oxidation of glycerol is investigated.
Experimental
Catalyst preparation
A monometallic gold catalyst and two bimetallic gold-platinum catalysts supported on 7-Al2O3 (Puralox SCCa 5150, Sasol Germany; SBET = 162 m2/g, Vpore = 0.47 cm3/g, dp < 250 μm) with a total metal loading of 2.2 wt.% were prepared.
Gold was loaded on to the support via direct anionic exchange. A solution containing 0.5, 0.2, and 0.1 g/l for the monometallic and the two bimetallic catalysts, respectively, and pH of approximately 4.5 was prepared from a HAuCl4 stock solution containing 250 g Au/l (Mintek). The pH of the solution was adjusted by the addition of a dilute NaOH solution. 10 g of alumina support was added to 1 litre of the gold-containing solution. The mixture was stirred (700 r/min) for 22 hours at room temperature. The gold-containing catalyst precursor was filtered off and washed with deionized water. The catalyst precursor was subsequently reslurried in 250 ml of an aqueous ammonia (25 wt.%) solution to remove residual chloride present in the catalyst. The catalyst precursor was filtered off after 20 minutes and washed with deionized water. Subsequently, the catalyst was dried in an oven at 110°C for 10 minutes and calcined in hydrogen at 200°C for 2 hours.
Slurry impregnation was used to deposit platinum on to the (gold-loaded) support. The required amount of tetraamine platinum tetrachloride monohydrate ((NH3)4PtCl2.H2O; Strem Chemicals) was dissolved in 50 ml of deionized water and added to the support. The solution was removed in a rotary evaporator operating at 80°C and 250 mbar. The catalyst was subsequently calcined in hydrogen at 200°C for 2 hours.
Catalyst characterization
The metal loading on the catalyst was determined using AAS after digesting a sample of the catalyst in HF/HCl. An initial estimate on the metal crystallite size distribution on the synthesized catalysts was obtained from transmission electron microscopy (TEM) images, recorded on a JEM 1200EXII (JEOL) operating at 120 kV. Energy dispersive X-ray analysis (EDX) was used to distinguish between gold and platinum crystallites.
Catalyst testing
The liquid phase glycerol oxidation was performed in a 500 ml three-necked round bottomed flask equipped with an overhead condenser at pH 10, a temperature of 60°C, and atmospheric pressure. Before each run, 4 ml of 0.3 M NaOH solution and 0.2 g of catalyst was added to 200 ml of deionized water. The flask was placed in a water bath and the solution was stirred at 400 r/min while maintaining the temperature at 60°C. Oxygen was sparged through the solution at a rate of 300 ml(NTP)/min. After 2 hours, 1 ml of a 6.84 M glycerol solution was injected into the solution marking the start of the reaction. The pH was controlled at pH 10 by titrating the solution with a 0.3 M NaOH solution using an auto-titrator (Tritoline Alpha plus). Samples (1 ml) of the reaction mixture were taken every 10 minutes and were immediately filtered to remove any catalyst from the sample.
The products of the liquid-phase glycerol oxidation were separated by HPLC using a Biorad column (Biorad Aminex HPX-87-H, 300 mm χ 7.8 mm) and an aqueous 0.01 M H2SO4 solution as the mobile phase (flow rate 0.6 ml/min) and detected with a RI detector and UV detector operating at 210 nm. The products were identified and their response factors were determined using reference compounds of the expected oxidation products.
Results and discussion
Table I shows an overview of the catalysts used in this study. The total metal loading in the supported catalysts was kept constant at 2.2 wt.%. Hence, the gold content in the bimetallic catalyst was reduced by 37 percent and 72 percent respectively.

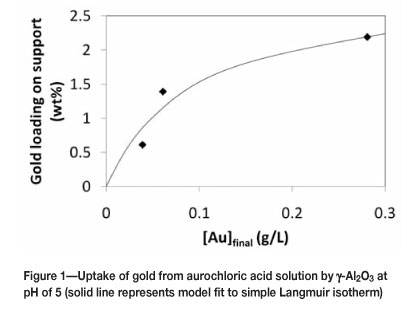
Gold was loaded onto the support via direct anionic exchange. The loading efficiency of gold onto alumina using direct anionic exchange from an aurochloric acid solution at pH of 5 ranged from 44-69 percent. The uptake of anionic gold complexes from the solution at pH of approximately 5 can be reasonable described using a Langmuir isotherm (see Figure 1). The maximum amount of gold loaded on this support would then correspond to approximately 2.8 wt.%, corresponding to 0.54 Au atoms per square nanometre, which is approximately 0.5 times the uptake reported for the uptake of PtCl62- on alumina (Regalbuto et cel., 1999). This difference can be explained in terms of the differences in the geometry of the adsorbed complexes.
A narrow gold crystallite size distribution is obtained via direct anionic exchange of aurochloric acid on to y-Al2O3 (see Figure 2). It is interesting to note that the average gold crystallite size in the monometallic gold catalysts is approximately 50 percent larger than that in the bimetallic gold- platinum crystallites. The gold uptake during the synthesis of the monometallic gold catalyst represents approximately 75% of the maximum uptake of the gold chloride complex by the support, whereas the gold uptake during the synthesis of the bimetallic gold-platinum catalysts represents less than 50 percent of the maximum uptake. It might be speculated that the high gold density may lead to some sintering in the following heat treatment(s).
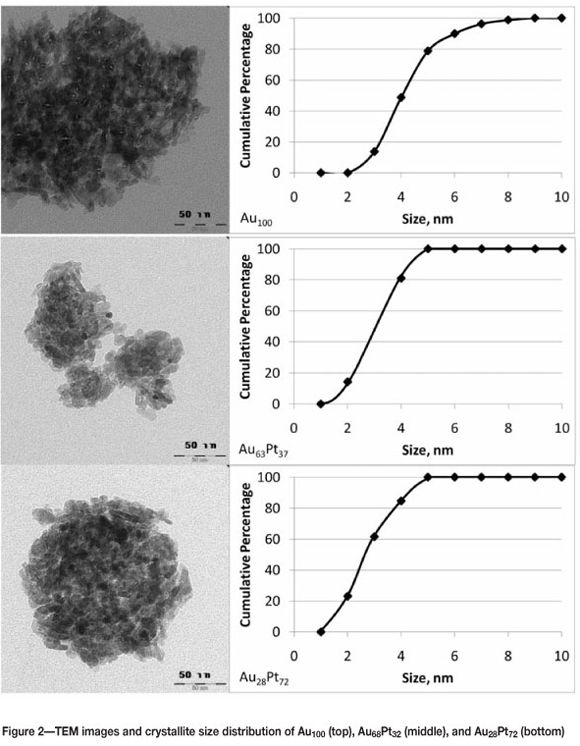
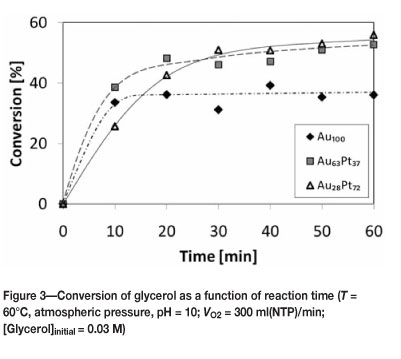
Figure 3 shows the glycerol conversion as a function of the reaction time. The monometallic gold catalyst, Au100, shows a conversion of approximately 34 percent after a reaction time of 10 minutes. The conversion of glycerol does not increase appreciably further after this reaction time-indicating severe and rapid deactivation of the catalyst. The bimetallic catalyst, Au63Pt37, shows after a reaction time of 10 minutes a slightly higher conversion of approximately 39 mol%, which increases rather slowly to approximately 55 mol% after 1 hour. The slow increase in the conversion followed by the initial high conversion indicates that this catalyst deactivated, but less severely than the monometallic gold catalyst. The bimetallic gold-platinum catalyst with the lowest gold loading, Au28Pt72, showed the lowest initial conversion of approximately 26 mol% after a reaction time of 10 minutes. The conversion of glycerol increases steadily up to 30 minutes, after which the conversion increases only slightly, again indicating deactivation of the catalyst. The two bimetallic catalysts reached final conversions that were close to each other.
The turnover frequency for the glycerol conversion (based on the total number of moles of metal in the reactor) of the alumina supported catalysts are in the same range as those reported for Au supported on MgAl2O4 spinels (Villa et al., 2010), for Au-Pt on carbon (Dimitratos et al., 2006a), and for Au-Pd on carbon (Dimitratos et al., 2006b). It should further be noted that the average gold crystallite size in the monometallic gold catalyst was only 50 percent larger than that in the bimetallic gold-platinum catalysts. Hence, the turnover frequency expressed per surface gold atom decreases with increasing platinum content in the catalyst, if it can be assumed that the gold surface area is adequately represented by the average crystallite size of the gold crystallites detected by TEM.
The deactivation in liquid-phase reactions might be ascribed to sintering, leaching, metal oxidation, poisoning, or by formation of oligomeric/polymeric species (Besson and Gallezot, 2003). Leaching is typically observed via re-use of the spent catalyst and was observed for Au/CeO2 (Demirel et cl. , 2007b), although it is known that leaching from alumina support is more facile than from, for example, carbon support materials (Besson and Gallezot, 2000). Metal oxidation is not expected in noble metal catalysed oxidation reactions. Hence, possible mechanisms for catalyst deactivation are sintering, (self-)poisoning by product compounds, and deposition of oligomeric species on the surface. The initially more active monometallic gold catalyst is more prone to deactivation than the bimetallic Au63Pt37 catalyst, which in turn deactivates faster than the bimetallic Au28Pt72 catalyst. The monometallic gold catalyst has on average larger gold crystallites than the bimetallic Au-Pt catalysts. Hence, the monometallic gold catalyst is expected to deactivate the least, if sintering (or leaching) is the dominant deactivation mechanism. Self-poisoning by product compounds of 'over-oxidation' (Besson and Gallezot, 2000) could be the cause of the observed deactivation order, since more active catalysts are then expected to deactivate faster.
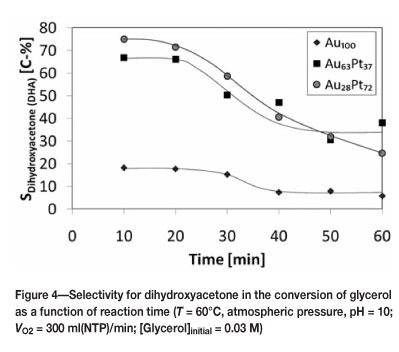
Dihydroxyacetone was the main product observed in the oxidation of glycerol (see Figure 4). It should be noted that the low selectivity for dihydroxyacetone observed with the monometallic gold catalyst is caused by the large amount of coke obtained with this catalyst, thus substantiating the high rate of deactivation obtained with this catalyst (vide supra). The selectivity for dihydroxyacetone decreases gradually with time, indicating the secondary conversion of dihydroxyacetone. Dihydroxyacetone is thought to form from the base catalysed tautomerisation of the primary oxidation product glyceraldehyde. However, it can be inferred by extrapolating the selectivity of dihydroxyacetone to a reaction time of zero that other products (and in particular glyceric acid) are also formed as primary products in the oxidation of glycerol. This can be seen in Figure 5.

The selectivity of glyceric acid increases slowly with time, as can be seen in Figure 5. The same trend is observed for glycolic acid. This, together with the fact that selectivity to dihydroxyacetone decreases over the same time is in accordance with the proposed mechanism wherein glyceric and glycolid acid forms as secondary products upon further oxidation of dihydroxyacetone (Ketchie et al. 2007).
Tartronic acid was also identified in the product samples but was found to have a selectivity of less than 10 percent and is not shown in Figure 4. The four products identified appear to be the main products from the oxidation reaction over the AuPt catalysts. A carbon balance performed over the course of the reaction confirms this, as can be seen in Figure 6. The missing carbon is less than 10 percent for both bimetallic catalysts. There is approximately 30 percent carbon unaccounted for with Au100. This explains the greater deactivation of the monometallic catalyst, since the missing carbon is most likely coke. The bimetallic catalysts, having less gold and therefore a slower deactivation, are not prone to the same degree of coke formation.

Conclusions
This study focuses on an alternative, environmentally friendly way of producing useful chemicals from a renewable source, i.e. the heterogeneously catalysed oxidation of glycerol under atmospheric pressure and mild temperature and controlled pH conditions with oxygen as the oxidizing agent. Alumina-supported gold catalysts were prepared by anionic ion exchange and some impregnated with platinum. The activity and selectivity in the oxidation reaction of the monometallic gold catalyst was compared to that of the bimetallic gold-platinum catalysts. The gold loading on the catalysts prepared by ion exchange was determined by atomic absorption spectroscopy. Platinum was then impregnated to achieve a total metal loading equal to the highest gold loading. TEM images were obtained and analysed to obtain average gold crystallite size.
The bimetallic catalysts achieve a 20 percent higher conversion in the oxidation reaction as compared to monometallic gold. The main products achieved with the bimetallic catalysts were dihydroxyacetone, glyceric acid, glycolic acid, and tartronic acid. These products, together with the unconverted glycerol, make up 90 percent of the carbon in the system. For monometallic gold, the unconverted glycerol and the above four products identified make up only 70 percent of the total carbon in the system, indicating the possible formation of coke. The bimetallic catalysts displayed a greater selectivity to dihydroxyacetone, glyceric acid, and glycolic acid as compared to monometallic gold. No significant activity was observed with the monometallic gold catalyst beyond 10 minutes and the glycerol conversion stayed constant, indicating deactivation of the catalyst. The bimetallic catalysts displayed a slower rate of deactivation and the carbon balance confirmed that it is likely that less coke was formed. In the catalysts investigated, it was found that the fraction of platinum in the bimetallic catalyst has no significant effect on conversion and selectivity.
References
Besson, M. and Gallezot, P. Selective oxidation of alcohols and aldehydes on metal catalysts. Catalysis Today, vol. 57, 2000. pp. 127-141. [ Links ]
Besson, M. and Gallezot, P. Deactivation of metal catalysts in liquid phase organic reactions. Catalysis Today, vol. 81, 2003. pp. 547-559. [ Links ]
Bianchi, C.L., Canton, P., Dimitratos, N., Porta, F., and Prati, L. Selective oxidation of glycerol with oxygen using mono and bimetallic catalysts based on Au, Pd and Pt metals. Catalysis Today, vol. 102-103, 2005. pp. 203-212. [ Links ]
Carrettin, S., McMoru, P., Johnston, P., Griffin, K., Kiely, C.J., and Hutchings, G.J. Oxidation of glycerols using supported Pt, Pd, and Au catalysts. Physical Chemistry Chemical Physics, vol. 5, 2003. pp. 1329-1336. [ Links ]
Demirel, S., Lehnert, K., Lucas, M., and Claus, P. Use of renewable for the production of chemicals: Glycerol oxidation over carbon supported gold catalysts. Applied Catalysis B: Environmental, vol. 70, 2007a. pp. 637-643. [ Links ]
Demirel, S., Kern, P., Lucas, M., and Claus, P. Oxidation of mono- and ployal-cohols with gold: comparison of carbon and ceria supported catalysts. Catalysis Today, vol. 122, 2007b. pp. 292-300. [ Links ]
Demirel-Gülen, S., Lucas, M., and Claus, P. Liquid phase oxidation of glycerol over carbon supported gold catalysts. Catalysis Today, vol. 103, 2005. pp. 166-172. [ Links ]
Dimitratos, N., Messi, C., Porta, F., Prati, L., and Villa, A. Investigation on the behavior of Pt(0)/carbon and Pt(0),Au(0)/carbon catalysts employed in the oxidation of glycerol with molecular oxygen in water. Journal of Molecular Catalysis A: Chemical, vol. 256, 2006b. pp. 21-28. [ Links ]
Dimitratos, N., Sanchez-Lopez, J.A., Lennon, D., Porta, F., Prati, L., and Villa, A. Effect of particle size on monometallic and bimetallic (Au,Pd)/Con the liquid phase oxidation of glycerol. Catalysis Letters, vol. 108, 2006b. pp. 147-153. [ Links ]
Ketchie, W.C., Murayam, M., and Davis, R.J. Promotional effect of hydroxyl on the aqueous phase oxidation of carbon monoxide and glycerol over support Au catalysts. Topics in Catalysis, vol. 44, 2007. pp. 307-317. [ Links ]
Ma, F. and Hanna, M.A. Biodiesel production: a review. Bioresource Technology, vol. 70, 1999. pp. 1-15. [ Links ]
Morrison, L.R. Glycerol. Kirk Othmer Encyclopedia of Chemical Technology. Wiley & Sons, New York, 2000. [ Links ]
Porta, F. and Prati, L. Selective oxidation of glycerol to sodium glycerate with gold-on-carbon catalyst: and insight into reaction selectivity. Journal of Catalysis, vol. 224, 2004. pp. 397-403. [ Links ]
Regalbuto, J.R., Navada, A., Shadid, S., Bricker, M.L., and Chen, Q. An experimental verification of the physical nature of Pt adsorption onto alumina. Journal of Catalysis, vol. 184, 1999. pp. 335-348. [ Links ]
Villa, A., Gaiassi, A., Rosetti, I., Bianchi, C.L., van Benthem, K., Veith, G.M., and Prati, L. Au on MgAl2O4 spinels: the effect of support surface properties in glycerol oxidation. Journal of Catalysis, vol. 275, 2010. pp. 108-116. [ Links ]
Zhou, C.-H., Beltramini, J.N., Fan, Y.-X., and Lu, G.Q. Chemoselective catalytic conversion of glycerol as a biorenewable source to valuable commodity chemicals. Chemical Society Reviews, vol. 37, 2008. pp. 527-549. [ Links ]
Zope, B.N., Hibbitts, D.D., Neurock, M., and Davis, R.J. Reactivity of the gold/water interface during selective oxidation catalysis. Science, vol. 330, 2010. pp. 74-78. [ Links ]
©The Southern African Institute of Mining and Metallurgy, 2012. SA ISSN2225-6253. This paper was first presented at the ZrTa2011 New Metals Development Network Conference, 12-14 October 2011, Mount Grace Country House & Spa, Magaliesburg.

