










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Industrial Engineering
versión On-line ISSN 2224-7890
versión impresa ISSN 1012-277X
S. Afr. J. Ind. Eng. vol.28 no.3 Pretoria 2017
http://dx.doi.org/10.7166/28-3-1841
GENERAL ARTICLES
An investigation into the value of a standardised global pharmacovigilance reporting system
M. Schurer*; L. Bam; I.H. de Kock
Health Systems Engineering and Innovation Hub, Department of Industrial Engineering, Faculty of Engineering, Stellenbosch University, South Africa
ABSTRACT
Pharmacovigilance (PV) is based on the medical assessment of adverse medical events or drug-related problems, collected within organised health programmes. The large number of different PV systems, the equally large number of stakeholders within such systems (pharmaceutical companies, government regulatory bodies, national and international clinical regulatory bodies, healthcare workers, etc.), and the significant number of dimensions along which the effectiveness and efficiency could be measured, adds to this complexity. Furthermore, the lack of a standardised reporting protocol across the various PV systems hinders efforts to manage PV coherently on a global scale. This paper proposes the value of a standardised global PV reporting system by evaluating the systemic effects of the lack of such a standardised system.
OPSOMMING
Farmakologiese waaksaamheid (PV) is gebaseer op die mediese assessering van nadelige mediese gebeure of dwelmverwante probleme wat deur georganiseerde gesondheidsprogramme versamel word. Die groot aantal verskillende PV-sisteme, die ewe groot aantal belanghebbendes in sulke stelsels (farmaseutiese maatskappye, regerings, nasionale en internasionale kliniese reguleringsliggame, gesondheidswerkers, ens.), en die aansienlike aantal dimensies waarvolgens die effektiwiteit en doeltreffendheid gemeet kan word, dra by tot hierdie kompleksiteit. Verder belemmer die gebrek aan 'n gestandaardiseerde rapporterings-protokol wat oor die verskillende PV-sisteme strek die pogings om PV op 'n wêreldwye skaal konsekwent te bestuur. Hierdie artikel stel die waarde van 'n gestandaardiseerde globale PV rapporteringsprotokol voor deur die sistematiese gevolge van die gebrek aan so 'n gestandaardiseerde stelsel te evalueer
1 INTRODUCTION
No medication is inherently safe: each treatment situation is unique, and each patient can respond differently to a specific treatment. The pharmacodynamics of biologics and chemically synthesised drugs on a molecular level are numerous and intricate; and it is resource-intensive to fully characterise the interactions of a drug in vitro. The topic of drug safety was brought to the world's attention when, in the late 1950s, thousands of babies in the United Kingdom were born with congenital deformities (phocomelia) after in utero exposure to the seemingly safe drug Thalidomide. Thalidomide was originally marketed in 1957 as a sedative, but was also prescribed for pregnant women to alleviate the symptoms of morning sickness that are associated with the early stages of pregnancy. The World Health Organization (WHO) established the International Drug Monitoring Programme (IDMP) in 1968, in direct response to the Thalidomide tragedy [1].
Pharmacovigilance (PV) is based on the medical assessment of adverse medical events or drug-related problems, collected within organised health programmes. Within these programmes it is vital to be able consistently to identify the nature of events, their severity, and their likelihood of occurrence, and to assess causality in connection with the suspected drug(s) or medicine(s). An adverse drug reaction (ADR) is defined as being any undesirable effect of a medication beyond its intended therapeutic effect [2]. PV is a responsibility that is shared by all stakeholders in the health system, from pharmaceutical multinationals and regulatory agencies to healthcare professionals (HCPs) and the patients themselves.
A PV system can be understood as consisting of an ADR reporting mechanism, data collation, causality analysis, risk assessment, decision-making, deliberated design and implementation of the appropriate action, and the evaluation of the outcomes achieved [3].
Standardisation is a principal tool used in quality improvement initiatives that focus on cost reduction and on identifying and eliminating inefficiencies within systems [4]. The concept of standardisation is broad, and spans multiple domains [5]. The standardisation of a system must be performed by subject matter experts from each of the various domains included in the system, along with a wide variety of stakeholders who have a vested interest in the system.
This paper aims to connect the potential benefits of a standardised reporting system with the challenges brought about by the fragmented nature of the current global PV context. A review of the literature on ADR reporting shows that the traditional method of spontaneous reporting is not effective, and gives rise to a variety of problems such as poor data quality and insufficient data capturing due to the under-reporting of ADRs by both HCPs and patients.
2 CONTEXT
For a medicine or therapeutic drug to gain approval from clinical regulatory bodies and to receive marketing authorisation, it must meet stringent requirements and pass multiple stages of pre-clinical and clinical trials. On completion of these trials, the regulatory body conducts a comprehensive review of the drug in question, to assess its potential benefits and harms. Once a drug has been approved and the marketing authorisation has been granted, the drug may be marketed within the jurisdiction governed by the relevant regulatory body.
When a drug is marketed on a global scale, the number of patients using the drug can be in the order of millions. This presents an opportunity for a robust global PV system to receive large amounts of data from new patient groups through the spontaneous reporting of ADRs. With a large and multi-ethnic1 population consuming medications, identifying new drug interactions is made more feasible. The consumption of a medication across a large population would also allude to the effects of consumption at different dosages and via different administration routes (intravenous, inhalation, oral, etc.).
The current global PV landscape is complex. A simplified view can be seen in Error! Reference source not found.. The Uppsala Monitoring Centre (UMC), founded in 1978 by the WHO to support the Programme for International Drug Monitoring, coordinates PV activities among 127 member countries. It facilitates the development of PV systems in 28 associate member countries.
The UMC manages and maintains the ADR database, VigiBase, which receives ADR reports from all the WHO member countries. In 2015, the VigiBase database had about 11 million ADR reports from 120 countries [6]. The International Society for Pharmacovigilance (ISoP in Figure 1) plays a supportive role in providing educational and administrative advice to many pharmaceutical manufacturers and many countries with PV systems. Two notable regulatory authorities are the FDA (Food and Drug Administration) of the US, which makes use of the FDA Adverse Event Reporting System (FAERS); and the EMA (European Medicines Agency), which manages the Eudra-Vigilance reporting system. Countries that fall outside the regulatory jurisdiction of the FDA and EMA typically have their own Departments of Health (DoH) and respective National Regulatory Authorities (NRA) that co-ordinate PV activities on a national level.
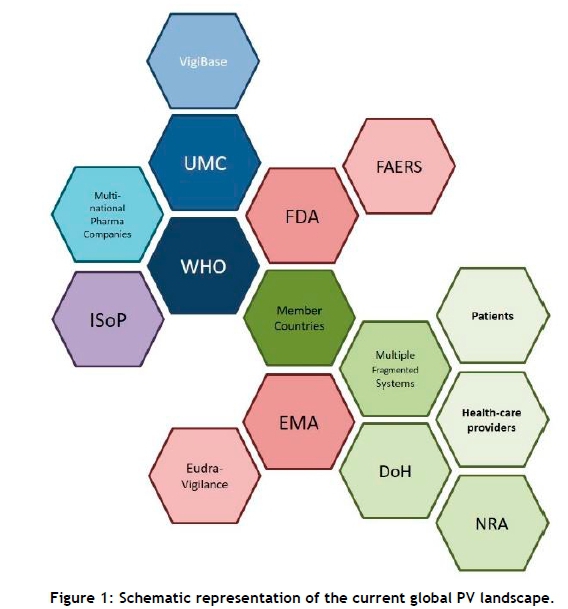
The fragmented nature of PV in the global context presents the UMC with a multitude of challenges. Currently, problems such as under-reporting and the communication of incomplete, unrepresentative, and uncontrolled data prove to be significant barriers to detecting and characterising new adverse drug interactions and ADRs [7]. A 2016 study by Bailey et al. [8] identified 108 ADR reporting systems and highlighted a number of challenges associated with the lack of a standardised ADR reporting form.
3 REPORTING METHODS AND THE EFFECTS OF A LACK OF STANDARDISATION
Data management is a key principle of pharmacovigilance. Sources of data include non-clinical and clinical trials, the scientific literature, pharmacoepidemiologic studies, and spontaneous reporting systems. Pharmacovigilance reporting systems rely primarily on the generation and detection of signals - that is, the communication of an ADR or adverse drug event (ADE) made by a patient, manufacturer, or healthcare provider to the appropriate PV centre. The unsolicited or spontaneous reporting of such ADRs is the cornerstone of data generation in post-marketing drug safety and surveillance. However, research suggests that spontaneous reporting is not a sufficiently comprehensive method of generating the data needed to make quantitative conclusions about the safety of medicines in the long term ([9],[10],[11]).
3.1 Reporting methods
There are three primary methods of reporting ADRs and drug safety information. The first is spontaneous reporting (SR). The other two are active surveillance methods: cohort event monitoring (CEM) and targeted spontaneous reporting (TSR).
3.1.1 Spontaneous reporting
Spontaneous reporting during the post-marketing phase generates the majority of drug safety data, even more so than the clinical trials during the drug development process [12]. Lester et al. [12] found that between 52 and 55 per cent of drug label changes were the result of spontaneous reporting; this demonstrates the significance of spontaneous reporting in PV activities. Spontaneous reporting involves the unsolicited generation of a signal by a healthcare provider or a patient relating to the suspicion of an ADR. This method is better than active surveillance methods because it incurs little or no administrative cost; it covers a large population of potential reporters and a large profile of drugs; and it allows for the monitoring of a medicine throughout its entire life cycle.
3.1.2 Cohort event monitoring
Cohort event monitoring (CEM) involves the prospective study of the ADRs associated with a specific drug within a small group of patients - a cohort. The primary benefit of CEM is realised when it is used to observe the effects of a new medicine in the early stages of post-marketing authorisation [10]. Although all PV activities are centrally focused on patient safety, CEM focuses on a specific medication for the time before and during the control period.
3.1.3 Targeted spontaneous reporting
Targeted spontaneous reporting (TSR) is a methodology similar to spontaneous reporting, but involving well- defined patient groups where healthcare professionals (HCPs) are on the lookout for specific ADRs [10]. TSR is an active method of surveillance in a well-defined population group, whereas spontaneous reporting is a passive method of surveillance used within an undefined population [13]. Being an active surveillance method, TSR is more resource-intensive than spontaneous reporting. However, TSR produces reporting data of a higher standard. TSR methods have shown strong potential in low- and middle-income countries for the monitoring of drug safety over extended periods of time in populations with specific disease burdens, such as HIV and TB [14].
3.2 Effects of a lack of standardisation
A brief summary of all the issues related to the lack of standardisation is presented here. The likely impact of standardisation on the various stages of the PV reporting system will be discussed in the next section. The most widespread challenge facing PV is a high level of under-reporting ([15],[16],[17],[18],[12]), typically attributed to a lack of knowledge, time, and incentive ([19],[16],[20]), which alludes to the lack of standardised reporting protocols and methodologies. PV also faces socio-cultural challenges, such as the existence of a culture of fear surrounding the reporting of ADRs due to a fear of undue disciplinary action being taken against HCPs ([21],[22],[19]). The literature also highlights the urgent need for educational awareness about PV activities and the simplification of the ADR reporting process to improve public participation ([23],[24],[25],[11]). Accountability among all stakeholders in a health system is important for the overall successful functioning of the system. A systematic review of ADR reporting systems by Bailey et al. [8] found a high degree of variability, with 1,782 distinct data elements having been identified across 108 systems. This shows that ADR report forms need to have standardised terminology and a comprehensive set of unique data elements.
3.3 What is meant by a standardised reporting system?
Given the scope of this research, a standardised reporting system is envisioned to have the following characteristics:
-
A global system in which an ADR is reported once, with data of high quality to facilitate causality analysis;
-
A transparent system in which data is accessible by all stakeholders, including public health programmes (PHPs), regulatory authorities (RAs), manufacturers, HCPs, patients, and the public at large;
-
A system that ensures the confidentiality of patients;
-
A system that reduces the fragmentation and duplication of data and resources;
-
A system that improves resource use in resource-limited contexts;
-
A system that reduces administrative pressure, allowing HCPs to direct their attention to other priorities and giving them more time to report ADRs; and
-
A system that enables HCPs to make more informed therapeutic decisions and improve patient safety.
The objectives of this system would be:
-
To reduce the frequency and severity of ADRs by widening the scope of pharmacovigilance on a global level;
-
To improve causality analysis and risk assessment, allowing HCPs to make more informed therapeutic decisions;
-
To enable quantitative conclusions to be made about the safety of medicines for long-term use; and
-
To improve the communication of drug safety information between HCPs and patients;
4 ANALYSIS, AND THE EFFECTS OF THE LACK OF A STANDARDISED GLOBAL PV REPORTING SYSTEM
ADRs are a significant cause of morbidity, mortality, and increasing costs for PHPs [26]. By facilitating the communication and collation of comprehensive ADR data, the objectives of PV reporting can be achieved. These objectives include characterising known reactions, measuring risk, identifying new reactions by detecting signals, characterising drug interactions, identifying risk factors such as age, gender, dosage etc., assessing safety in various patient groups (pregnancy, elderly, paediatric, etc.), and detecting and measuring the inefficacy of medicines.
Data pooling has a direct effect on the accuracy of ADR frequency estimation. By not pooling data from across the international landscape, we lessen the rate of detection of rare but clinically significant ADRs [27], specifically those that occur with a low incidence rate but that can pose a significant threat to public and patient safety. By pooling data, these silent but serious ADRs can be more readily detected.
Patel et al. [28] found that the mean preventable ADRs leading to hospitalisation was 45.11 per cent, with the primary suspects being cardiovascular system drugs (28.1 per cent), nonsteroidal antiinflammatory drugs (NSAIDs) (16.1 per cent), nervous system drugs (16.9 per cent), and musculoskeletal drugs (16.1 per cent). Inappropriate drug selection due to misdiagnosis, toxic drug serum levels, and failure to predict or avoid known drug interactions are among the leading causes of preventable ADRs.
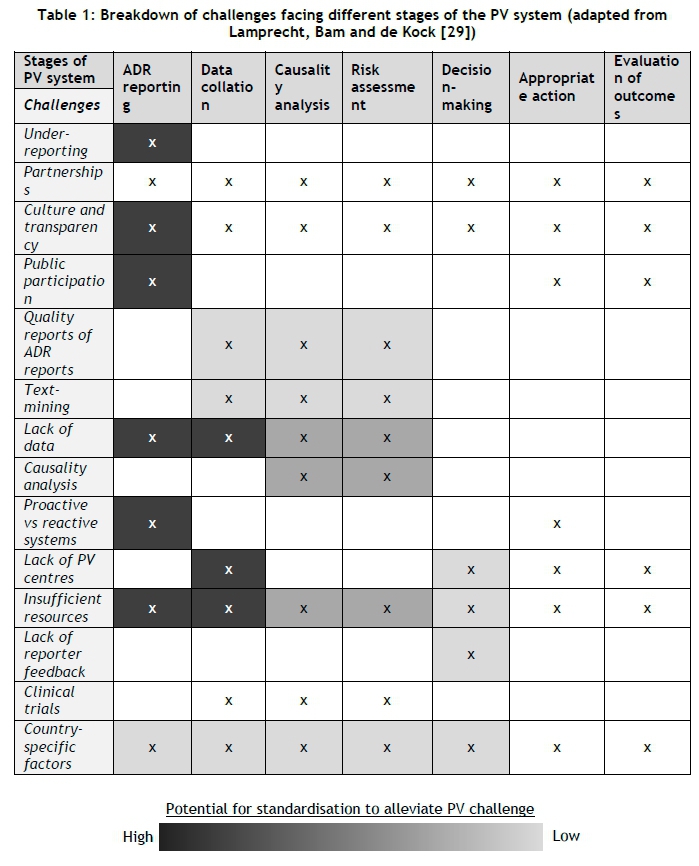
A multitude of complex challenges hinder pharmacovigilance activities. Table 1 shows a breakdown of the challenges, and the part of the PV system with which they are associated [29]. The colour grading scale (added to the table as part of this research on the value of standardising ADR reporting) shows the extent to which a standardised ADR reporting system would alleviate the respective challenges. The colour gradings are explained and motivated in the subsections below.
It is important to consider the limitations of spontaneous reporting systems. Due to the unsolicited nature of spontaneous reporting, problems such as under-reporting, the inability to derive incidence and prevalence rates due to the lack of denominator data, and the potential for reports to contain insufficient clinical data are widespread.
4.1 Under-reporting
A systematic review performed by Hazell and Shakir [17] provided evidence of widespread underreporting across 12 countries, and stated that the rate of under-reporting was as high as 94 per cent. The notion of under-reporting in the literature has often been attributed to the knowledge, attitudes, and practices [23] of healthcare providers. Common concerns and barriers to reporting by healthcare providers have been identified. These include lack of knowledge of the functioning of a PV system, lack of incentive to report ADRs, lack of time, and, interestingly, a fear of blame [21]. Healthcare providers, particularly in low-income countries, have been found to be reluctant to report ADRs, as they believe it reflects poorly on their professional ability to treat their patients [16]. A standardised reporting system would assist in restoring confidence in both healthcare providers and patients by ensuring the confidentiality of submitted reports.
Through standardising the PV reporting system and associated protocols, PV in general will become easier to include, and more manageable, in all undergraduate training curricula of HCPs. The concept of a minimum requirement report is worth further investigation. This form would seek to capture the most important characteristics of the ADR, of the medication, and of the patient. If a more detailed follow-up report must be filed, the HCP and the patient would be notified accordingly. HCPs often cite a lack of time as a primary reason for not filling out ADR reports; a minimum requirement form would be less time-consuming to complete, and could therefore improve underreporting rates. Additional reflections on the expected impact of a minimum reporting form on resource utilisation follows in Section 4.6.
A further improvement would be the use of a feedback mechanism to provide the HCP and patient with an acknowledgement of receipt of the ADR report. The provision of feedback to reporters of ADRs would almost certainly increase the overall rate of spontaneous reporting among HCPs and patients alike. This feedback could comprise a simple acknowledgement of receipt, or could provide information to the reporter about an appropriate course of action to take in order to treat the symptoms of the ADR experienced by the patient.
4.2 Culture and transparency
There is a need for a change in culture and transparency in ADR reporting [30]. There is a misconception among many healthcare practitioners, particularly those in low- and middle-income countries ([23],[31]), that pharmacovigilance is not the responsibility of a public health programme, but rather that of the pharmaceutical industry itself. Transparency is improving in PV systems around the globe, with some countries (such as Canada and the Netherlands) making their spontaneous reporting databases freely accessible to the public [32]. It is important to understand the distinction between transparency and confidentiality, and how they do not necessarily contradict one another.
A transparent system would enable all stakeholders to interact with the system and to extract all the ADR data that pertains to their role in the PV system. Confidentiality of patient information can be achieved by not disclosing information to unauthorised parties. In the PV context, there are certain elements of an ADR report that should be disclosed to all stakeholders, but there are some that contain sensitive information about the patient. By ensuring the anonymity of patients when sharing their ADR report data, they can be confident that the generated data will not be used against them by any third party.
Standardisation would improve the transparency of the system and improve the accessibility of up-to-date information about the safety of medicines. Improving accessibility to the latest information would allow patients to have a greater degree of confidence in their PHP. Accountability can be improved through improving the knowledge, attitudes, and practices of HCPs during their undergraduate training. When HCPs understand that the responsibility of PV is shared by all stakeholders in the health system, they will be able to report ADRs, confident that their actions will not result in undue disciplinary action being taken against them.
4.3 Public participation
Patient-centricity is a key aspect of an effective PV system. Through fostering an environment in which the patient is well-informed and confident, public participation is likely to improve. PHPs need to provide accurate and transparent information to the public to gain public trust; a notable example of this is with national immunisation programmes. For the public to cooperate with a national programme for immunisation, the risks and benefits of the programme must be adequately communicated.
An electronic web-based form should be made available to all members of the health system, and could be available in all languages to improve ease of use. As mentioned in Section 4.1, the use of a feedback mechanism to provide HCPs and patients with acknowledgement of receipt of their ADR report would likely improve levels of reporting. People will be more likely to participate in the system when they are made to feel valued by the system. Giving the patient a platform to make their voice heard and acknowledging their report is an important step in improving public participation. The encouragement of patient reporting would increase the rate of overall spontaneous reporting, and would likely enable earlier detection of unexpected ADRs [33].
4.4 Lack of data
Developing a standardised ADR spontaneous reporting framework would directly support the WHO's efforts to manage pharmacovigilance activities on a global scale. An effective ADR spontaneous reporting system would allow for the early detection of ADRs in the post-marketing phase. Incidence rates of the ADRs could be established, and the identification and characterisation of novel drug interactions could take place. Bailey et al. [8] found a high degree of variability among the 108 reporting systems in their study. They also pointed to the lack of standardised data elements in reporting forms, having identified 1,782 distinct data elements, which were mapped to 33 reporting concepts. A standardised reporting form would include a comprehensive list of unique data elements, prompting the extraction of all demographically and clinically relevant data relating to the patient and the ADR.
This reduction in the number of data elements can be achieved through the use of standardised terminology. The two principal medical terminology directories are the Medical Dictionary for Regulatory Activities (MedDRA) and the World Health Organization Adverse Reactions Terminology Dictionary (WHO-ART). Discrepancies between MedDRA and WHO-ART contribute to the problem of data quality. A standardised reporting system would allow for improved causality analysis through the pooling of uniform data, extracted from a comprehensive set of unique data elements using standardised terminology.
4.5 Proactive vs reactive system
The unsolicited nature of spontaneous reporting makes it a reactive system. With 55 per cent of all drug label changes in the United States since 2010 being in direct response to spontaneous reporting of ADRs [12], it is clear that an improved reporting system can benefit proactive drug safety activities such as drug labelling and package inserts. By improving efficiency in ADR reporting, drug safety labels will reflect the latest safety information, allowing HCPs to make more informed therapeutic decisions.
4.6 Lack of PV centres and insufficient resources
Healthcare systems are unique to the countries in which they function. Pharmacovigilance activities, particularly in low- and middle-income countries (LMIC), are often considered 'nice to have', and take second place to efforts focused primarily on improving access to medicines [6]. Given the variety and complexity of public healthcare programmes around the world, the UMC faces considerable challenges and difficulties in attempting to co-ordinate the IDMP. A study by Olsson et al.[34] found that only 41 per cent of the countries studied had any budget allocated to pharmacovigilance.
The lack of standardisation results in reporting systems receiving reports with incomplete or insufficient data. This places stress on the system and diverts resources such as time, people, and money away from other more important activities [20]. Accurately quantifying the cost of healthcare is difficult enough on a national public health programme level; assessing the costs associated with ADRs on a global level is even more challenging. However, research has been conducted to expose the reasons for these costs and to give estimates of the costs incurred by PHPs.
Costs associated with ADRs are attributed to extended lengths of hospitalisation, the cost of treating illnesses caused by ADRs, and the cost of avoiding ADRs. Most of the literature on the pharmacoeconomic aspects of ADRs focuses primarily on the costs of the hospitalisation of patients due to ADRs. A 1998 study found that complications from ADRs account for the fourth to sixth leading cause of death in hospitalised patients in the United States of America [35]. Total hospital costs associated with ADRs in the US have been estimated to be at least $30 billion, and as high as $130 billion, annually.
By pooling resources among PHPs and drug manufacturers, a mechanism could be established to facilitate the follow-up of minimum requirement reports that have been received, as required. If sufficient minimum requirement reports, at an aggregate level, generate a signal that alludes to the presence of an unexpected ADR, then the appropriate resources can be allocated for a more thorough investigation into the safety of a specific medicine, such as a Cohort Event Monitoring initiative.
5 COMPONENTS AND MINIMUM REQUIREMENTS OF A STANDARDISED GLOBAL PV REPORTING SYSTEM
The World Health Organization provides guidelines and a significant number of resources for conducting PV activities within PHPs. The minimum requirements for a functional PV system were first described in 2010 [36] and include, among other elements, a national spontaneous reporting system with an ADR reporting form.
A standardised global PV reporting system (along with the standardised reporting form, such as the form described by Bailey et al. [8]) would rely heavily on the political will of the participating countries (together with the support of national regulatory agencies) to provide a clear legal basis and to inform all stakeholders of their relevant roles and responsibilities. Another critical success factor is the availability of stable funding, technical capability, and human resources for PV activities within the PHPs of the participating countries.
The WHO cites the greatest challenge and responsibility of national PV centres within the public health programme to be the effective and open communication of drug safety information between the NRAs and the HCPs and patients [1]. The Erice Declaration of 1998 [1] serves as a framework to ensure that all stakeholders across the PV system act according to the highest ethical, professional, and scientific standards when communicating drug safety information. The Erice Declaration states the following:
-
Drug safety information must serve the health of the public.
-
Education in the appropriate use of drugs, including interpretation of safety information, is essential for the public at large, as well as for health care providers.
-
All the evidence needed to assess and understand risks and benefits must be openly available.
-
Every country needs a system with independent expertise to ensure that safety information on all available drugs is adequately collected, impartially evaluated, and made accessible to all.
-
Innovation in drug safety monitoring needs to ensure that emerging problems are promptly recognised and efficiently dealt with, and that information and solutions are effectively communicated.
Informed HCPs and patients are encouraged to take up stewardship in carrying out PV activities and adhering to standards to ensure the successful communication of drug safety information.
6 CONCLUSION
Pharmacovigilance activities contribute to the prevention of unnecessary patient harm, improved clinical practice, and the support of research and education activities. Patient safety remains the central focus of all PV activities, with the end goal of assisting healthcare providers to make more informed therapeutic decisions for their patients.
In order to identify and characterise evidence-based causal relationships between ADRs and their suspected medicines, standardised data must be communicated efficiently and effectively at every stage of the PV system. The standardised reporting system investigated in this research must be defined by experts and key stakeholders in the PV system. A thorough stakeholder analysis must be performed so that the perspectives of those stakeholders with a vested interest in the outcomes of the system - i.e., the regulators, healthcare providers, patients, etc. - are not neglected.
The development of a standardised ADR spontaneous reporting framework would directly support the WHO's efforts to manage pharmacovigilance activities on a global scale. If regional differences are insuperable, or a universal solution is not found to be feasible, the sharing of best practices, together with the leveraging of PV capacity and capability through collaboration and partnership, must be considered. Governments and their respective regulatory bodies need to provide a political mandate to all street-level bureaucrats, together with supporting legislation and standard operating procedures (SOPs), to improve awareness of, and adherence to, good PV practices across all levels of their public health programmes (PHPs).
A collaborative global effort is needed to fast-track PV development around the world. Those countries without the necessary facilities, expertise, or resources for PV need them the most [31].
Developing countries often have the highest disease burden on their PHPs. The strength of global pharmacovigilance lies in the integration of various national PV systems. Although efforts may have been made to standardise parts of the PV system, the focus must shift to the diffusion and successful adoption and implementation of those standards.
REFERENCES
[1] World Health Organization. 2002. The importance of pharmacovigilance: Safety monitoring of medicinal products. Geneva, Switzerland: World Health Organization. [ Links ]
[2] Pirmohamed, M., Breckenridge, A.M., Kitteringham, N.R. and Park, B.K. 1998. Fortnightly review: Adverse drug reactions. British Medical Journal, 316(7140), p.1295. [ Links ]
[3] Strengthening Pharmaceutical Systems (SPS). 2009. Supporting pharmacovigilance in developing countries: The systems perspective. Submitted to the US Agency for International Development by the SPS Program. Arlington: Management Sciences for Health. [ Links ]
[4] Blind, K. and Mangelsdorf, A. 2016. Motives to standardize: Empirical evidence from Germany. Technovation, 48, pp.13-24. [ Links ]
[5] Xie, Z., Hall, J., McCarthy, I.P., Skitmore, M. and Shen, L. 2016. Standardization efforts: The relationship between knowledge dimensions, search processes and innovation outcomes. Technovation, 48, pp.69-78. [ Links ]
[6] Olsson, S., Pal, S.N. and Dodoo, A. 2015. Pharmacovigilance in resource-limited countries. Expert review of clinical pharmacology, 8(4), pp.449-460. [ Links ]
[7] Banerjee, A.K., Okun, S., Edwards, I.R., Wicks, P., Smith, M.Y., Mayall, S.J., Flamion, B., Cleeland, C. and Basch, E. 2013. Patient-reported outcome measures in safety event reporting: PROSPER consortium guidance. Drug safety, 36(12), pp.1129-1149. [ Links ]
[8] Bailey, C., Peddie, D., Wickham, M.E., Badke, K., Small, S.S., Doyle-Waters, M.M., Balka, E. and Hohl, C.M. 2016. Adverse drug event reporting systems: A systematic review. British Journal of Clinical Pharmacology, 82(1), pp.17-29. [ Links ]
[9] Ndagije, H., Nambasa, V., Namagala, E., Nassali, H., Kajungu, D., Sematiko, G., Olsson, S. and Pal, S. 2015. Targeted spontaneous reporting of suspected renal toxicity in patients undergoing highly active antiretroviral therapy in two public health facilities in Uganda. Drug Safety, 38(4), pp.395-408. [ Links ]
[10] Pal, S.N., Duncombe, C., Falzon, D. and Olsson, S. 2013. WHO strategy for collecting safety data in public health programmes: Complementing spontaneous reporting systems. Drug Safety, 36(2), pp.75-81. [ Links ]
[11] Layton, D. and Shakir, S.A. 2015. Specialist cohort event monitoring studies: A new study method for risk management in pharmacovigilance. Drug Safety, 38(2), pp. 153-163. [ Links ]
[12] Lester, J., Neyarapally, G.A., Lipowski, E., Graham, C.F., Hall, M. and Dal Pan, G. 2013. Evaluation of FDA safety-related drug label changes in 2010. Pharmacoepidemiology and Drug Safety, 22(3), pp. 302-305. [ Links ]
[13] Mehta, U., Dheda, M., Steel, G., Blockman, M., Ntilivamunda, A., Maartens, G., Pillay, Y. and Cohen, K. 2014. Strengthening pharmacovigilance in South Africa. South African Medical Journal, 104(2), pp.104106. [ Links ]
[14] Pal, S.N., Olsson, S. and Brown, E.G. 2015. The monitoring medicines project: A multinational pharmacovigilance and public health project. Drug Safety, 38(4), pp.319-328. [ Links ]
[15] Koutkias, V.G. and Jaulent, M.C. 2015. Computational approaches for pharmacovigilance signal detection: Toward integrated and semantically-enriched frameworks. Drug Safety, 38(3), pp.219-232. [ Links ]
[16] Graham, J.E., Borda-Rodriguez, A., Huzair, F. and Zinck, E. 2012. Capacity for a global vaccine safety system: The perspective of national regulatory authorities. Vaccine, 30(33), pp.4953-4959. [ Links ]
[17] Hazell, L. and Shakir, S.A. 2006. Under-reporting of adverse drug reactions. Drug Safety, 29(5), pp.385-396. [ Links ]
[18] Marques, F.B., Penedones, A., Mendes, D. and Alves, C. 2016. A systematic review of observational studies evaluating costs of adverse drug reactions. ClinicoEconomics and Outcomes Research, 8, p.413. [ Links ]
[19] Hasford, J., Goettler, M., Munter, K.H. and Müller-Oerlinghausen, B. 2002. Physicians' knowledge and attitudes regarding the spontaneous reporting system for adverse drug reactions. Journal of Clinical Epidemiology, 55(9), pp.945-950. [ Links ]
[20] Dal Pan, G.J. 2014. Ongoing challenges in pharmacovigilance. Drug Safety, 37(1), pp.1-8. [ Links ]
[21] Kim, J., Kim, S., Jung, Y. and Kim, E.K. 2010. Status and problems of adverse event reporting systems in Korean hospitals. Healthcare Informatics Research, 16(3), pp.166-176. [ Links ]
[22] Suku, C.K., Hill, G., Sabblah, G., Darko, M., Muthuri, G., Abwao, E., Pandit, J., Osakwe, A.I., Elagbaje, C., Nyambayo, P. and Khoza, S. 2015. Experiences and lessons from implementing cohort event monitoring programmes for antimalarials in four African countries: Results of a questionnaire-based survey. Drug Safety, 38(11), pp.1115-1126. [ Links ]
[23] Bhagavathula, A.S., Elnour, A.A., Jamshed, S.Q. and Shehab, A. 2016. Health professionals' knowledge, attitudes and practices about pharmacovigilance in India: A systematic review and meta-analysis. PloS One, 11(3), p.e0152221. [ Links ]
[24] Sevene, E., Mariano, A., Mehta, U., Machai, M., Dodoo, A., Vilardell, D., Patel, S., Barnes, K. and Carné, X. 2008. Spontaneous adverse drug reaction reporting in rural districts of Mozambique. Drug Safety, 31(10), pp.867-876. [ Links ]
[25] Stergiopoulos, S., Brown, C.A., Felix, T., Grampp, G. and Getz, K.A. 2016. A survey of adverse event reporting practices among us healthcare professionals. Drug Safety, 39(11), pp. 1117-1127. [ Links ]
[26] Nazer, L.H., Eljaber, R., Rimawi, D. and Hawari, F.I. 2013. Adverse drug events resulting in admission to the intensive care unit in oncology patients: Incidence, characteristics and associated cost. Journal of Oncology Pharmacy Practice, 19(4), pp.298-304. [ Links ]
[27] Olsson, S. 1998. The role of the WHO programme on international drug monitoring in coordinating worldwide drug safety efforts. Drug Safety, 19(1), pp.1-10. [ Links ]
[28] Patel, N.S., Patel, T.K., Patel, P.B., Naik, V.N. and Tripathi, C.B. 2017. Hospitalizations due to preventable adverse reactions: A systematic review. European Journal of Clinical Pharmacology, 73(4), pp.1-14. [ Links ]
[29] Lamprecht, I.V.B., Bam, L. and de Kock, I.H. 2017. An investigation into the prospects of existing technologies to address the challenges faced by pharmacovigilance systems, in Taking up stewardship - The 28th Annual SAIIE Conference 2017 Proceedings, Vanderbijlpark, South Africa, October 25-27, 2017. [ Links ]
[30] Chruscicki, A., Badke, K., Peddie, D., Small, S., Balka, E. and Hohl, C.M. 2016. Pilot-testing an adverse drug event reporting form prior to its implementation in an electronic health record. SpringerPlus, 5(1), p.1764. [ Links ]
[31] World Health Organization. 2006. Pharmacovigilance an essential tool. Geneva: World Health Organization. [ Links ]
[32] Molokhia, M., Tanna, S. and Bell, D. 2009. Improving reporting of adverse drug reactions: Systematic review. Clinical Epidemiology, 1, p.75. [ Links ]
[33] Jarernsiripornkul, N., Patsuree, A. and Krska, J. 2017. Public confidence in ADR identification and their views on ADR reporting: Mixed methods study. European Journal of Clinical Pharmacology, 73(2), pp.223231. [ Links ]
[34] Olsson, S., Pal, S.N., Stergachis, A. and Couper, M. 2010. Pharmacovigilance activities in 55 low-and middle-income countries. Drug Safety, 33(8), pp.689-703. [ Links ]
[35] Lazarou, J., Pomeranz, B.H. and Corey, P.N. 1998. Incidence of adverse drug reactions in hospitalized patients: A meta-analysis of prospective studies. The Journal of the American Medical Association, 279(15), pp.1200-1205. [ Links ]
[36] World Health Organization. 2010. Minimum requirements for a functional pharmacovigilance system. Geneva: World Health Organization. [ Links ]
[37] Maggo, S.D., Savage, R.L. and Kennedy, M.A. 2016. Impact of new genomic technologies on understanding adverse drug reactions. Clinical Pharmacokinetics, 55(4), pp.419-436. [ Links ]
* Corresponding author 16497457@sun.ac.za
1 Different ethnic groups could respond differently to certain medications, due to the effects of slight variations in their DNA, known as single nucleotide polymorphisms [37].


{kind=link}