










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Psychiatry
versão On-line ISSN 2078-6786
versão impressa ISSN 1608-9685
S. Afr. j. psyc. vol.21 no.1 Pretoria Fev. 2015
http://dx.doi.org/10.7196/sajp.573
ARTICLE
When apparent schizophrenia is excluded
H F FourieI; L KoenIII; D J H NiehausIV; M SchoemanII; U A BothaV
IMB ChB; Department of Psychiatry, Faculty of Medicine and Health Sciences, Stellenbosch University, Cape Town, South Africa
IIMSc (Med) (Genet Counsel), BSc (Hons) (Genet); Division of Molecular Biology and Human Genetics, Faculty of Medicine and Health Sciences, Stellenbosch University and Tygerberg Hospital, Cape Town, South Africa
IIIMB ChB, MMed (Psych), FCPsych, PhD; Department of Psychiatry, Faculty of Medicine and Health Sciences, Stellenbosch University, Cape Town, South Africa
IVMB ChB, MMed (Psych), DMed, FCPsych, PhD; Department of Psychiatry, Faculty of Medicine and Health Sciences, Stellenbosch University, Cape Town, South Africa
VMB ChB, MMed (Psych), FCPsych; Department of Psychiatry, Faculty of Medicine and Health Sciences, Stellenbosch University, Cape Town, South Africa
ABSTRACT
Where must a clinician turn when straightforward schizophrenia suddenly turns out to be just the opposite? Fortunately, these days, there are protocols for just about everything. But how much value do these add? This article outlines the journey of our attempt to follow one such protocol.
In the beginning
Mr R was a 30-year-old male who was diagnosed with schizophrenia at the age of 19, after he presented with a typical combination of disorganised behaviour and persecutory delusions, against a background of daily comorbid polysubstance abuse. Owing to a pattern of frequent readmissions and poor treatment adherence, he was included into the Assertive Community Treatment (ACT) service at Stikland Hospital (SH) in Cape Town.
His mother (Mrs H), aged 54, was receiving treatment at the local clinic for major depressive disorder (MDD) and hypertension at the time. Shortly after her son's inclusion into the ACT service, she had her first admission to SH in January 2008, 3 years after she had first presented to the clinic. A diagnosis of MDD with psychotic features was made (persecutory delusions and auditory hallucinations). During this admission, no extrapyramidal side-effects were seen, but falls on two separate occasions were reported (attributed to blood pressure effects of her newly initiated antipsychotics). After discharge, Mrs H defaulted on her medication almost immediately, and 3 months later the mental health nurse referred her back to SH with a history of having presented with psychotic features. The referral letter also noted that the client had 'numerous bruises resulting from falls and bumping into things'. Mrs H was assessed at SH, found to be mildly psychotic with akathisia, but not in need of admission. Propranolol was prescribed for the akathisia and she was also included into the ACT service. As compliance on oral medication remained an obstacle, the depot was reboarded.
On a home visit, the ACT keyworker noted that Mrs H had worsening extrapyramidal side-effects (rigidity and akathisia), and arranged for her to be seen by the ACT consultant psychiatrist. She was found to have rigidity in all limbs, brisk reflexes and a bizarre gait with heel-toe stepping and pronation of both feet. She was also very restless and struggled to cooperate during the examination. The first available neurology outpatient appointment was in 4 months' time at Tygerberg Hospital. Her typical antipsychotics were stopped and she was started on oral risperidone, with a view to switching her to the risperdal long-acting injectable.
Mrs H missed the first appointment with neurology and was only seen in November 2009. She was subjected to a battery of tests and bloodwork, and booked for an MRI; all this was part of the standard work-up for difficult-to-diagnose movement disorders. Her working diagnosis remained that of severe side-effects owing to typical antipsychotics. Both the neurology notes and psychiatry notes clearly stated that no athetoid movements were noted on examination. Subsequently, the MRI in May 2010 showed only nonspecific changes.
Finally, in July 2010, at a follow-up, Mrs H tested positive for Huntington's disease.
Educating ourselves
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder that exhibits a varying prevalence across ethnic groups. It is estimated to affect 0.01% of the white population,[1] and a recent meta-analysis indicated an increased prevalence in Europe, North America and Australia in comparison with Asian countries.[2] The HD mutation is located on the Huntington gene (HTT), on the short arm of chromosome 4. HTT contains a trinucleotide repeat of cytosine-adenine-guanine (CAG) base pairs. In normal subjects, the number of repeats is usually between 10 and 26, but in HD the CAG sequence is repeated >36 times, forming a mutant strand of Huntington (mHtt). The exact mechanism by which the altered form of the protein leads to the pathological changes in HD has not yet been determined, but it is known to target the medium spiny neurons of the brain.
Over the clinical course of the illness, HD patients typically exhibit impairment across the triad of cognitive, neurological and psychiatric spectra. Neurologically, the so-called Huntington's chorea (writhing type movements of the limbs and other parts of the body) is the most common presentation. This seems to develop owing to the loss of gamma-aminobutyric acid (GABA) inhibitory neurons in the caudate nucleus and the globus pallidus, while cognitively, HD patients typically develop subcortical dementia over time owing to the loss of acetylcholine-secreting neurons in the cerebral cortex.[3] The psychiatric spectrum is more varied than the cognitive, neurological and psychiatric spectra, with ~35 - 75% of HD patients developing symptomatology at some stage of the disease.[4-6] Most commonly, patients present with personality changes such as irritability, aggression and apathy, while depression has a lifetime prevalence of 39%.[7] In contrast to popular belief, the psychiatric symptoms may precede the onset of the movement disorder or dementia, and in fact, HD patients often present to psychiatric services first.
The typical age of onset is in the late thirties to early forties, but a juvenile variant is also well described (also known as Westphal variant HD or akinetic-rigid HD). The number of repeats of the CAG trinucleotide has been shown to inversely correlate with the age of onset of symptoms; the more repeats the parent has, the more unstable the replication process becomes, the higher the chance that the child will inherit a longer expansion with a possible earlier onset of symptoms (genetic anticipation).
The next step
Guidelines for the molecular genetics predictive test in Huntington's disease were published in 1994 after the introduction of direct mutation analysis.[8] These were subsequently used by the University of Cape Town to instruct the development of the predictive testing protocol that has been implemented in the Western Cape Province. According to the protocol, predictive testing is considered when a person is asymptomatic and has a family history of confirmed HD. However, when a patient does show signs or symptoms indicative of the onset of this disease, predictive testing no longer applies; rather, testing is done for diagnostic purposes.
The rest of this article focuses on the ease of use of such a protocol in a developing world setting, using our own experience as an illustration. Difficulties encountered during this process, suggestions for possible improvements as well the ethical considerations that arise from managing such clients will form part of the discussion. Written consent was obtained from all participants mentioned and the final manuscript was also submitted to the Stellenbosch University Health Research Ethics Committee.
Diagnostic v. predictive testing
After the diagnosis of Mrs H, the option of genetic testing was discussed with Mr R as he was psychiatrically stable. He subsequently tested positive for Huntington's disease, with 44 CAG repeats in comparison with his mother's 42.
In a patient (such as Mr R) who presents with psychosis, making a decision as to the applicability of predictive testing could pose an ethical dilemma. Does this patient present with schizophrenia as a separate disease process or are the symptoms related to the pathological changes of HD? If the answer to the former is yes, then the patient should be regarded as asymptomatic (also termed presymptomatic) and he/she should proceed with the process of counselling for predictive testing according to the protocol. If the symptoms are judged to possibly be part of the presentation of HD, the patient is regarded as symptomatic and the tests could be done diagnostically.
Available data suggest that 5 - 25% of HD patients will present with psychotic features,[1,7] and that the likelihood of such a presentation is increased when the HD patient has a family member with HD who also presented with psychosis.[7]
Three main theories exist as to why some HD patients present with psychosis while others do not: (i) genetically, these patients have a different subtype of HD,[7] possibly linked to the length of the CAG repeats;[9,10] (ii) cooccurrence in a 'vulnerable' brain[7] and (iii) developmental effect due to the stress of growing up in a disrupted family structure secondary to the presence of serious illness, of which one has a 50% chance of developing.[11-13]
Currently, although we are unable to distinguish clinically between the psychosis of HD and schizophrenia, there are various possible pointers. A recent review on secondary psychosis suggests that several questions can be asked to determine whether the psychosis is as a result of a general medical condition (GMC).[14] These include questions on atypicality of presentation as well as temporal and/or physiological relationship to a GMC and/or substances. Additionally, primary illness factors such as age of onset, family history, outcomes and characteristic findings on special investigations should always be considered.
It could be argued that there is no immediate benefit for the patient presenting with psychosis alone to know that he/she will develop HD, as the main psychopharmacological management will not differ. Therefore, the predictive rather than diagnostic testing option should be followed. However, early diagnosis could have advantages. Choice of antipsychotic is important, as both haloperidol and risperidone have been shown to slow down the progression of the striatal dysfunction in HD clients.[15-17] Multidisciplinary team intervention and family support (including predictive testing) as well as access to resources not available within the mental health system can be offered.
In the case of Mr R, he had a parent with confirmed HD, was deemed to be symptomatic and was therefore tested after informed consent was obtained. In spite of numerous post-test counselling sessions, Mr R's insight remained very limited. Seen in overview, in all likelihood his ability to make an informed decision about predictive testing was significantly impaired.
Involving the rest of the family
A family meeting with the treating doctor was arranged where Mrs H's mother (Mrs SG) was present, and based on her information, a genogram was constructed (Fig. 1).
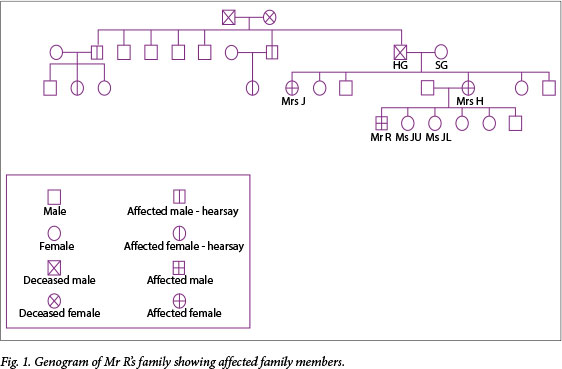
Mrs SG gave the history that her husband (Mr HG) had a condition ('the clumsy disease') that he developed later in his life. According to her, he was diagnosed with a form of seizure disorder. Another one of her daughters, Mrs J, was admitted to an old-age home at a young age with a diagnosis of Parkinson's disease, and Mrs H believed she also had 'the clumsy disease'.
Mrs J was visited and found to display the typical choreiform movements of HD. She was also at an advanced stage of dementia and, regarding her as HD symptomatic, we obtained informed consent from her legal guardian for diagnostic testing; she tested positive with 43 CAG repeats.
Attempting predictive testing
A family meeting with the genetic counsellor was planned; as per the protocol a genetic counsellor from Tygerberg Hospital was requested to facilitate the session in order to ensure provision of adequate information on HD and psychosocial support.
The family had no telephone and contact could only be established via home visits, done jointly by the ACT keyworker and doctor. Flyers bearing the details of the meeting were also left at their home.
In an attempt to make it more accessible, the meeting was held at the local community healthcare centre. Only Mrs H's five children attended this meeting. Only two of the five siblings were eligible for predictive testing as the other three were under the age of 18 years (according to the protocol the age of consent is 18); Taking into account that the teenage pregnancy rate in South Africa (SA) is 54/1 000 (WHO/Global Health Observatory Data Repository[18]), this decision should likely be revisited as family planning is vital in HD.
The second meeting with both eligible siblings was scheduled just over 2 months after the initial meeting. Mrs JU had married a few months prior to the meeting and was pregnant, while Ms JL was unmarried but had a 2-year-old daughter. Mrs JU had discussed predictive testing with her husband and, even though he encouraged her to do so, decided against testing. Her reasons for doing so were mainly that she thought that she wouldn't be able to handle a positive result and the 'constant waiting' for the symptoms to appear. Ms JL decided to go ahead mainly to determine if her child was also at risk, but also because she 'would rather know than not know'. A follow-up appointment was arranged with Ms JL for an individual session. She was also asked to identify someone as a support partner (as per the protocol).
Ms JL did not attend the follow-up at the end of January 2012, indicating that she could not get off from work. Another session could only be arranged for early in April as the genetic counsellor was unavailable. Once again, Ms JL failed to attend. The counsellor then made contact and explicitly asked if she had doubts about testing; she affirmed this. Further counselling and predictive testing was thus put on hold, pending notice from Ms JL.
Barriers to effective protocol implementation in a developing world setting
Resources
Futter et al.[19] concluded that factors such as low income and education level contribute to poor access to predictive testing centres and programmes in the Western Cape. The challenges experienced during our process are not unique to HD clients but are specifically frustrating when one is trying to work within the confines of a structured protocol. The family did not have a telephone or access to electronic communication so they could only be reached by means of the ACT team performing home visits. Obviously, this resulted in delays in arranging follow-up appointments. In addition, the family also made use of public transport and could not always attend meetings owing to the lack of funds or unpredictability of the transport system.
Genetic counsellors
According to the international protocol, a multidisciplinary team should be involved in the process of predictive testing, including a neurologist, clinical psychologist and a genetic counsellor. The counsellor's role is to inform the client about the disease, its inheritance pattern and therefore the client's risk of HD, to give as much information as possible about the advantages and disadvantages of predictive testing and to prepare them for the implications and outcome of the result, taking into consideration their psychosocial circumstances. The process is supposed to create an environment where the client has sufficient information, time and support to make an informed decision about predictive testing.
Currently, there are only 18 registered genetic counsellors (of which 3 are interns) in SA. According to the Genetic Service Plan for SA, the suggested genetic counsellor-to-person ratio is 1:500 000, which translates to a recommended >100 genetic counsellors in SA and >11 counsellors for the Western Cape. At present, there are only two genetic counsellor posts available in state practice in the Western Cape, both of which are part-time contract posts (4 days/week). The Gauteng Province has four permanent posts and KwaZulu-Natal one. In comparison, the USA has 2 500 registered genetic counsellors.
It would therefore come as no surprise that the genetic counsellor helping us had very limited availability. Adding to that the practical difficulties experienced communicating with the family, one has a clearer picture as to why the whole process was stretched over such an extended period of time.
Coordination of services
In most parts of the country, HD clients do not have easy access to coordinated services. Mr R's treating doctor was based at SH, the genetic counsellor at Tygerberg Hospital and the family had to be seen at the local community health centre in their area. The examination room used for this was not really designed to host a therapeutic intervention. Certainly these factors contributed to the difficulty in keeping the family engaged in the process.
Conclusion
The HD predictive testing programme in SA has previously been scrutinised, with numerous suggestions made to try to overcome some of the identified difficulties.[19] Our experience of the process once again highlighted many frustrations. Lack of some basic resources made adherence to goals of the protocol virtually impossible. It was very difficult to try to maintain a balance between giving the client enough time to process information, contemplate and make an informed decision about testing, and keeping the client engaged to prevent him/her from becoming lost to follow-up.
More posts for properly trained genetic counsellors are vital, and ideally, designated teams should be readily accessible at primary care level. One possible solution would be the implementation of mobile clinics.
Ultimately the protocol should serve to make the process as meaningful as possible to every client, and thus it should be more adaptable to the client's unique situation and needs.
References
1. Corrêa BB, Xavier M, Guimarães J. Association of Huntington's disease and schizophrenia-like psychosis in a Huntington's disease pedigree. Clin Pract Epidemiol Ment Health 2006;2:1. [http://dx.doi.org/10.1186/1745-0179-2-1] [ Links ]
2. Pringsheim T, Wiltshire K, Day L, Dykeman J, Steeves T, Jette N. The incidence and prevalence of Huntington's disease: A systemic review and meta-analysis. Mov Disord 2012;27(9):1083-1091. [http://dx.doi.org/10.1002/mds.25075] [ Links ]
3. Hall JE. Textbook of Medical Physiology. 12th ed. London: Saunders, 2011:694. [ Links ]
4. Dewhurst K, Oliver JE, Trick KLK, McKnight AL. Neuropsychiatric aspects of Huntington's disease. Confin Neurol 1969;31:258-268. [ Links ]
5. Folstein SE. Huntington's Disease: A Disorder of Families. Baltimore: John Hopkins University Press, 1989. [ Links ]
6. Cummings JL. Behavioural and psychiatric symptoms associated with Huntington's disease. Adv Neurol 1995;65:179-186. [ Links ]
7. Tsuang D, Almqvist EW, Lipe H, et al. Familial aggregation of psychotic symptoms in Huntington's disease. Am J Psychiatry 2000;157(12):1955-1959. [ Links ]
8. International Huntington Association and the World Federation of Neurology Research Group on Huntington's Chorea. Guidelines for the molecular genetics predictive test in Huntington's disease. Neurology 1994;44(8):1533-1536. [ Links ]
9. Kremer B, Goldberg P, Andrew SE, et al. A worldwide study of the Huntington's disease mutation: The sensitivity and specificity of measuring CAG repeats. N Engl J Med 1994;330(20):1401-1406. [ Links ]
10. Andrew S, Goldberg Y, Kremer B, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat Genet 1993;4(4):398-403. [ Links ]
11. Dewhurst K, Oliver J, McKnight A. Socio-psychiatric consequences of Huntington's disease. Br J Psychiatry 1970;116(532):255-258. [ Links ]
12. Enoch MA. The influence of gene-environment interactions on the development of alcoholism and drug dependence. Curr Psychiatry Rep 2012;14(2):150-158. [http://dx.doi.org/10.1007/s11920-011-0252-9] [ Links ]
13. Lucas-Thompson RG, Goldberg WA. Family relationships and children's stress responses. Adv Child Dev Behav 2011;40:243-299. [http://dx.doi.org/10.1016/B978-0-12-386491-8.00007-4] [ Links ]
14. Keshavan MS, Kaneko Y. Secondary psychoses: An update.World Psychiatry 2013;12(1):4-15. [http://dx.doi.org/10.1002/wps.20001] [ Links ]
15. Duff K, Beglinger LG, O'Rourke ME, Nopoulos P, Paulson HL, Paulson JS. Risperidone and the treatment of psychiatric, motor and cognitive symptoms in Huntington's disease. Ann Clin Psychiatry 2008;20(1):1-3. [http://dx.doi.org/10.1080/10401230701844802] [ Links ]
16. Johnston TG. Risperidone long-acting injection and Huntington's disease: Case series with significant psychiatric and behavioural symptoms. Int Clin Psychopharmacol 2011;26(2):114-119. [http://dx.doi.org/10.1097/YIC.0b013e3283407775] [ Links ]
17. Charvin D, Roze E, Perrin V, et al. Haloperidol protects striatal neurons from dysfunction induced by mutated huntingtin in vivo. Neurobiol Dis 2008;29(1):22-29. [http://dx.doi.org/10.1016/j.nbd.2007.07.028] [ Links ]
18. World Health Organization, and Global Health Observatory Data Repository. Global Health Observatory (GHO) data, South Africa: Country profiles. http://www.who.int/gho/countries/zaf/country_profiles/en/ (accessed 20 February 2013). [ Links ]
19. Futter MJ, Heckmann JM, Greenberg LJ. Predictive testing for Huntington's disease in a developing country. Clin Genet 2009;75(1):92-97. [http://dx.doi.org/10.1111/j.1399-0004.2008.01044.x] [ Links ]
 Correspondence:
Correspondence:
H F Fourie (hesterff@gmail.com)

