










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Psychiatry
versión On-line ISSN 2078-6786
versión impresa ISSN 1608-9685
S. Afr. j. psyc. vol.20 no.2 Pretoria abr./jun. 2014
http://dx.doi.org/10.7196/SAJP.494
CASE REPORT
Kleine-Levin syndrome as a neuropsychiatric presentation: A case report and review
A Lachman
MB ChB, FCPsych (SA), MMED (Psych), MPhil (Child Psych), Cert Child Psych (SA); Tygerberg Hospital, Cape Town; Department of Psychiatry, Stellenbosch University, Tygerberg, Cape Town, South Africa
ABSTRACT
Kleine-Levin syndrome (KLS) is a rare disorder characterised by hypersomnia, hyperphagia and behavioural disturbances. It typically occurs in adolescent men and may mimic a variety of neuropsychiatric conditions. Here we report what appears to be the first published case of KLS in South Africa and trace the clinical course, investigations and diagnostic difficulties encountered in the management of this condition. An overview of the literature guiding the diagnosis, differentials and treatment is discussed.
Kleine-Levin syndrome (KLS) is considered an exceptionally rare,[1] recurrent condition characterised by hypersomnia, hyperphagia and behavioural disturbances including hypersexuality, depression, irritability and derealisation.[1,2] It occurs typically in adolescent men, who are often misdiagnosed with depression, bipolar disorder, psychosis or seizure disorders. Patients are asymptomatic between attacks and return to baseline, with normal or nonspecific results of investigations. The disorder is at the interface of neurology and psychiatry, and no objective biochemical, neuropathological or structural imaging alterations have been identified. This paper describes a case of typical KLS, the first to be documented in South Africa (SA), and illustrates diagnostic difficulties in its presentation.
Case report
Over the course of 2 years, patient A presented to a tertiary hospital with six episodes of excessive sleepiness (hypersomnia) accompanied by blurred vision and headaches. These episodes were followed by changes in appetite, irritability of mood and periods of hypersexuality. His first presentation to hospital was in 2011, but on further enquiry the family reported similar episodes beginning in 2008 (at the age of 10 years), where he would complain of headaches and blurred vision, followed by deep sleep, sometimes in excess of 15 hours, before returning to his normal level of activity.
In 2011, at the age of 14 years, patient A was admitted to a psychiatric ward following an episode of aggression, hypersexuality and running away from home. A week before his admission he reported not sleeping well and being stressed while preparing for examinations. He was observed to be disorientated and fluctuating in his mental state, with periods of poor arousability. Investigations for delirium included two normal electroencephelograms (EEGs), a normal computed tomography (CT) scan of the brain, and a negative toxicology screen for illicit substances. He was empirically treated with sodium valproate for a seizure disorder and his aggression and hypersexuality improved with an atypical antipsychotic (risperidone). After discharge, he continued to function well at school but was readmitted with a second episode of aggression, hypersomnia, headaches and confusion in January 2012. This time he was treated for a postictal psychosis, which settled on an increased dose of risperidone. All investigations at this stage were normal. A week following discharge, he was readmitted with a relapse. This episode settled quickly with no adjustments in medication.
In July 2012, he presented again to a psychiatric ward following headaches, irritable mood, paranoia and blurred vision during his mid-year examinations at school. During this admission, haematological investigations and cerebrospinal fluid (CSF) examinations were all normal. An ophthalmological investigation was also normal. While in the ward, he developed swelling of the joints with restricted range of movement in both knee joints. After a review by a rheumatologist, a diagnosis of juvenile oligoarthritis was made, and he started receiving treatment with salazopyrine.
His combined diagnoses on discharge included a psychotic disorder due to a general medical condition, complex partial seizures and oligoarthritis. Outpatient appointments were regularly attended and the complaints varied from ongoing blurred vision to intermittent joint pain. His social and academic functioning remained intact. He had one episode of hypersomnia over a weekend, followed by a voracious appetite, but this resolved and he returned to school with no apparent cognitive fallouts.
On 1 January 2013, he began complaining of anxiety, with blurred vision, and became very distressed and clingy with his mother. An episode of sleepiness followed, which lasted 20 hours, together with a change in his appetite, after which he was brought to hospital. He reported feeling confused and sleepy, and presented as perplexed with an irritable mood during the interview. He was compliant with his medication regimen, which included sodium valproate, risperidone and salazopyrine. He settled quickly in the ward. All investigations, including a urine toxicology, were unremarkable. He was reviewed by the rheumatologist, an ophthalmologist and a neurologist and discharged with an appointment for a sleep EEG as an outpatient. An additional diagnosis of bipolar disorder not-otherwise-specified was entertained despite the lack of clear mood symptoms and no family history of mood or psychiatric disorders.
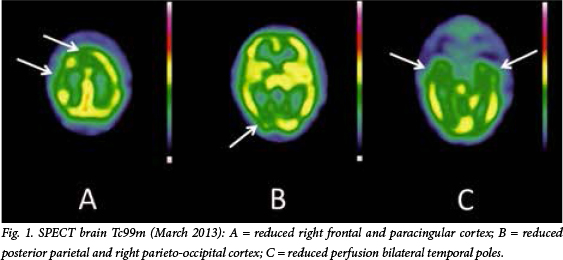
A month post discharge, he presented once again with an acute onset of hypersomnia (lasting 2 days) followed by emotional outbursts, including irritability, clingy behaviour, aggression and sexual disinhibition. He was fully compliant on medication, denied any substance abuse, and all investigations (including inflammatory markers) were normal. Subsequent investigations included a single photon emission CT (SPECT) of the brain (Fig. 1), which showed reduced perfusion to the right frontal, paracingulate posterior parietal and right parieto-occipital cortices. Perfusion was also reduced bilaterally to the temporal poles. A neurology consultation was requested and, following expert opinion, KLS was diagnosed.
He was discharged on only salazopyrine and risperidone at a low dose. However, his return to school was plagued by episodes of anxiety, poor concentration and memory impairment. A month later he was readmitted with hypersomnia, poor arousability and confusion. All investigations, including magnetic resonance imaging (MRI) of the brain, were normal. He was reviewed by rheumatology and medical genetics departments, who found examination unremarkable. Methylphenidate was initiated as a short course to help with the sleepiness. Despite resolution of the sleepiness, he remained irritable, intrusive, anxious and aggressive. There were no perceptual disturbances, but clear impairments in memory and attention.
Guided by anecdotal reports and cases documented in the literature,[2,3] he was given a trial of lithium carbonate after which his mood symptoms began to settle. A repeat SPECT of the brain showed no change to the areas of hypoperfusion. He is currently receiving individual educational facilitation to bridge him into mainstream schooling after a year of interrupted educational input.
Discussion
KLS diagnosis is difficult because the condition is seen infrequently and is often complicated by the presence of neurological and psychiatric symptoms.[4] The International Classification of Sleep Disorders (1990)[5] modified the diagnostic criteria for KLS and defined it as a syndrome composed of recurring episodes of undue sleepiness lasting for some days, which may or may not be associated with hyperphagia and abnormal behaviour.
Onset of the condition occurs typically in adolescence (81%) although younger onset of the first episode has been reported.[2] Aetiology and pathogenesis of KLS is unknown, but intermittent dysfunction in the hypothalamus is likely.[6] Inflammatory pro-cesses,[6] neurotransmitter (serotonin and dopamine) metabolism abnormalities[7] and viral infections[2] have all been suggested as possible contributors to KLS. Frequently, neurological conditions such as encephalitis, narcolepsy and circadian rhythm disorders may be considered in cases of KLS.[6,7]
Recorded precipitating factors for episodes include: upper respiratory tract infections, streptococcal infections, gastroenteritis, viral infections (chicken pox, Epstein Barr virus), substance use (cannabis and alcohol), head trauma and sleep deprivation or stress.[8] Our patient reported no stressors other than exam pressure and had a single episode of sleep deprivation preceding an attack. In addition, his only possible identifiable precipitant was considered to be the juvenile oligoarthritis.
Clinically, as demonstrated by this case, KLS may often be misdiagnosed and treated as bipolar disorder or a seizure disorder, despite there being an absence of evidence to support either diagnosis. Because of the cyclical nature of the presentation and disturbances in sleep and inhibitions, several authors have attempted to find a relationship between KLS and bipolar disorder.[6,8] The range of disturbed behaviours reported is wide, encompassing features suggestive of mild clouding of consciousness (disorientation), mood symptoms (irritability, dysphoria, impulsiveness, depression), overarousal/restlessness, disinhibition or psychotic features.[1,9] The striking presentation with behavioural disturbances warranting repeated investigations perhaps overshadowed the diagnosis in our patient, and the clue which lay in the symptom of repeated hypersomnia.
KLS is a diagnosis of exclusion, with symptoms that may mimic many more commonly occurring conditions that need to be explored first. In a systematic review of 186 case studies of KLS, Arnulf et al.[8] reported that most medical tests in KLS patients were aimed at eliminating epilepsy (EEG), focal brain lesions (brain imaging) and encephalitis (CSF analysis) as potential causes, and even when done during the acute episodes, results were unremarkable. Radiological investigations are usually normal, with routine MRI and EEG also being non-specific.[1] The most frequent reporting of abnormalities appear to be recorded on SPECT imaging[2] with reported perfusion deficits in the right or left thalamus, hypothalamus, basal ganglia, mesial temporal lobes and frontal lobe. [2,10] In our case, a SPECT done on patient A during an episode showed reduced perfusion to the right frontal, paracingular, posterior parietal, right parieto-occipital cortices and bilateral temporal poles. The interepisodic SPECT, however, showed no significant change to the pattern of perfusion deficits.
Evidence suggests that the course of KLS is often benign, with spontaneous improvement over several years although interepisodic morbidity especially resulting from mood disorders, and mild but long-lasting memory disturbances have been documented.[1,9] Episodes are less frequent towards the end of the disease course, and patients are considered cured only if they do not experience an episode for >6 years.[1,2] A high frequency of episodes in the beginning (as in patient A) is thought to be associated with a shorter duration of disease.[2]
Patients wish to alleviate the symptoms because of the crippling nature of the acute phase and the fact that episodes can stretch for weeks or months at a time, impairing their quality of life. Owing to the rarity of the disease, consistent reports or guidelines on treatment are sparse. There are no randomised, placebo-controlled trials of pharmacological treatment for KLS.[3] Drug trials of stimulants such as methylphenidate and amphetamine[2,8] have been suggested for symptomatic treatment during episodes of hypersomnia. Where psychotic symptoms are present, risperidone has been reported as most beneficial.[2,3] Preventative strategies have been suggested for use in individuals who experience >4 episodes a year.[2,3] Lithium has been the most frequently used treatment and reported as significantly beneficial in improving abnormal behaviour and reducing the duration of episodes while decreasing relapses.[3,11]
Patient A is currently largely asymptomatic on a therapeutic dose of lithium and risperidone, and only has complaints of minor memory difficulties at school. His last episode was 3 months prior to the initiation of lithium.
Conclusion
This report appears to be the first published case in SA of an adolescent with KLS presenting with neuropsychiatric symptoms to an acute psychiatric service. KLS is difficult to diagnose at first presentation because more serious neurological disorders need to be excluded. However, in adolescent males who present with periodic hypersomnia and psychiatric or behavioural disturbances with recurrent presentations yielding normal investigations, a diagnosis of KLS should be considered. The clinical manifestations as highlighted in this case emphasise the need for neurologists and psychiatrists to collaborate, especially in the management of diagnostically difficult adolescents.
Consent. Written consent was obtained from the adolescent and his parents for publication, and approved by the Human Research and Ethics Committee of Stellenbosch University, ethics reference number C13/07/002.
Acknowledgements. The author wishes to thank Profs Jonathan Carr of the Department of Neurology and James Warwick of the Department of Nuclear Medicine, Tygerberg Hospital, Stellenbosch University and Dr Eddy Lee-Pan of the Department of Neurology, University of Cape Town, for their assistance in managing the case and drafting this manuscript.
References
1. Ramdurg S. Kleine-Levin syndrome: Etiology, diagnosis and treatment. Ann Indian Acad Neurol 2010;13(4):241-246. [http://dx.doi.org/10.4103/0972-2327.74185] [ Links ]
2. Arnulf I, Floo TJ, Mignot E. Diagnosis, disease course and management of patients with Kleine-Levin Syndrome. Lancet Neurol 2012:11(10):918-928. [http://dx.doi.org/10.1016/S1474-4422(12)70187-4] [ Links ]
3. Oliveira MM, Conti C, Saconato H, Prado GF. Pharmacological treatment for Kleine-Levin syndrome. Cochrane Database Sys Rev 2013;8. [http://dx.doi.org/10.1002/14651858.CD006685.pub3] [ Links ]
4. Bonnet F, Thibaut F, Levillain D, Petit M. Kleine-Levin syndrome misdiagnosed as schizophrenia. Eur Psychiatry 1996;11(2):104-105. [http://dx.doi.org/10.1016/0924-9338(96)84788-2] [ Links ]
5. Thorpy MJ, Chairman, Diagnostic Classification Steering Committee. International Classification of Sleep Disorders: Diagnostic and Coding Manual. American Sleep Disorders Association, 1990. [ Links ]
6. Papacostas SS, Hadjivasilis V. Kleine-Levin syndrome: Report of a case and review of the literature. Eur Psychiatry 2000:15(4):231-235. [http://dx.doi.org/10.1016/S0924-9338(00)00239-X] [ Links ]
7. Cawthorn P. A disorder unique to adolescence? The Kleine Levin syndrome. J Adolesc 1990;13(4):401-406. [http://dx.doi.org/10.1016/0140-1971(90)90032-3] [ Links ]
8. Arnulf I, Zeitzer JM, File J, et al. Kleine-Levin syndrome: A systematic review of 186 cases in the literature. Brain 2005;128(12):2763-2776. [http://dx.doi.org/10.1093/brain/awh620] [ Links ]
9. Pike M, Stores G. Kleine-Levin syndrome: A cause of diagnostic confusion. Arch Dis Child 1994;71(4):355-357. [http://dx.doi.org/10.1136/adc.71.4.355] [ Links ]
10. Hong SB, Joo EY, Tae WS. Episodic diencephalic hypoperfusion in Kleine-Levin syndrome. Sleep 2006;29(8):1091-1093. [ Links ]
11. Muratori F, Bertini N, Masi G. Efficacy of lithium treatment in Kleine-Levin syndrome. Eur Psychiatry 2002;17(4):232-233. [http://dx.doi.org/10.1016/S0924-9338(02)00661-2] [ Links ]
 Correspondence:
Correspondence:
A Lachman
anusha@sun.ac.za

