










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSA Journal of Radiology
versão On-line ISSN 2078-6778
versão impressa ISSN 1027-202X
S. Afr. J. radiol. (Online) vol.21 no.1 Johannesburg 2017
http://dx.doi.org/10.4102/sajr.v21i1.1118
CASE REPORT
Monica S. Msomi; Nondumiso N. Dlamini
Department of Radiology, Pietermaritzburg Metropolitan Complex, College of Health Sciences, Nelson R. Mandela School of Medicine, University of KwaZulu-Natal, South Africa
ABSTRACT
Cherubism is rarely described in the African paediatric population. Orphanet currently lists cherubism as a rare disease; its prevalence is unknown and difficult to determine because of the wide clinical spectrum. Approximately 300 cases have been reported in various ethnic groups worldwide. This report analyses a child referred to our hospital for bilateral jaw swelling, diagnosed with cherubism based on clinical and radiological findings, and confirmed on histology.
Introduction
Cherubism is currently listed in Orphanet as a rare disease of unknown prevalence due to its wide clinical spectrum, with approximately 300 cases reported in various ethnic groups worldwide.1 It is an uncommon, benign, self-limiting fibro-osseous disorder characterised by painless progressive bilateral enlargement of the mandible and maxilla, first described by William Jones in 1933.2,3,4,5 Both hereditary and sporadic cases have been described.2,4,5 It is inherited as an autosomal-dominant disorder with variable penetrance, and a mutation in chromosome 4p16.3 has been demonstrated.2,4,6 Recent studies have shown it to be a genetically separate entity from fibrous dysplasia,2,4,5,6 of which it was initially thought to be a subset. It presents typically in childhood, as early as age 2,3,4,5 with painless symmetric enlargement of the jaw associated with slight upward turning of the eyes. Ancillary findings include dental arch and dental eruption abnormalities,2,3,5,6 and submandibular and cervical lymph node enlargement.3,6 Minor signs such as gum epulis and gum overgrowth are useful to diagnose relatives of affected children, in order to define a comprehensive family history. Extragnathic skeletal involvement is rare.2
Case report
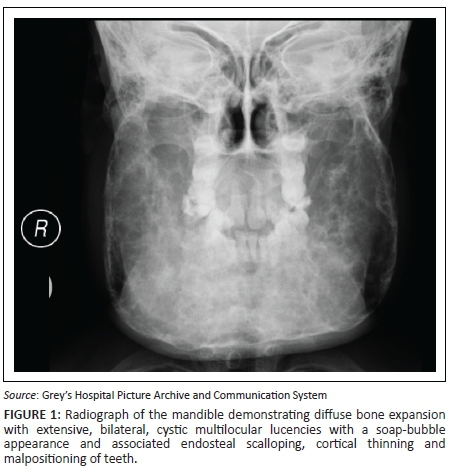
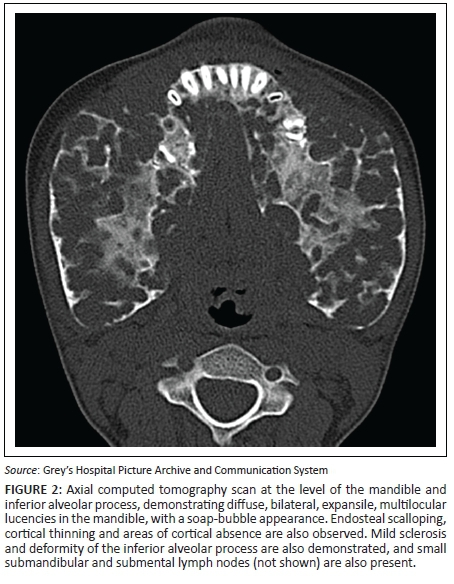
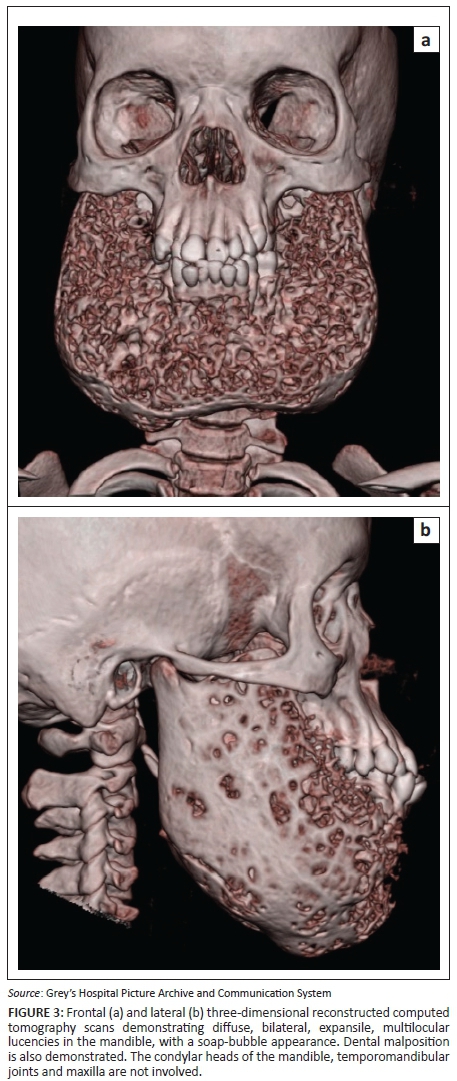
A 7-year-old girl presented with swollen gums and painless mandibular swelling for 2 years. The radiological findings demonstrated on a radiograph (Figure 1) and a computed tomography (CT) scan (Figures 2 and 3) of the mandible were in keeping with a diagnosis of cherubism. This was confirmed on histology. A definite family history was obtained, which ascertained a sporadic form of the disease. Genetic tests were not performed. Management was initially conservative, but the patient later had surgery for dental complications. A subsequent CT scan demonstrated static disease. The patient is currently undergoing periodical clinical follow-up.
Ethical consideration
The patient's identity was protected. This case report is purely for educational and academic purposes.
Discussion
The radiographic hallmark of cherubism is bilateral, symmetrical, multiloculated radiolucent lesions in the mandible extending from the region of the molar teeth towards the midline,5 resulting in a soap-bubble appearance of the jaw.3 Maxillary involvement is less frequent, characterised by a soft tissue density in the maxillary antrum resulting in the 'hard palate sign' on lateral skull radiographs.5 Absence of periosteal reaction is another important feature.2 Sparing of the mandibular condyles has traditionally been considered a hallmark of this condition, but there have been reports of involvement of the condyles.2,4,5 Unilateral cases of cherubism have been reported.3,5 Panoramic radiographs are acceptable for the initial diagnosis, but multi-planar and three-dimensional CT reconstructions are mandatory for optimal visualisation of the extent of disease.2 Histology has limited value for the diagnosis2 and demonstrates fibrous stroma containing abundant multinucleated giant cells,2,3,4,6 a finding which is not pathognomonic. The diagnosis, therefore, depends on clinico-radiological findings.2,3
Radiographic differential diagnoses for cherubism include cranio-facial fibrous dysplasia, Brown tumour of hyperparathyroidism, familial gigantiform cementoma,2,4 and central giant cell granuloma.3,5,6 Cherubism and fibrous dysplasia can be distinguished clinically and histologically. Features that favour the diagnosis of cherubism include bilateral mandibular involvement and limitation to the mandible. Furthermore, patients with fibrous dysplasia do not present with swollen cheeks or dental derangement4 and tend to present at a later age.5 Brown tumour is excluded on clinical and biochemical grounds. Lesions in central giant cell granuloma have a predilection to involve the anterior mandible, are rarely bilateral or symmetrical and tend to present later.5 Familial gigantiform cementomas consist predominantly of focal maxillary lesions often with extension into the orbits and nasal septum.2,4,5
Cherubism may be associated with other genetic diseases such as Ramon's syndrome, Noonan's syndrome, Jaffe-Campanacci syndrome and neurofibromatosis type 1.2,6 Our patient did not have any family history or express any clinical features of these conditions.
There is a tendency towards spontaneous remission,2,5 with partial or full regression and sclerotic involution by adulthood,2,4 mostly without the need for treatment.2,5,6 Medical agents may be used to reduce the size of lesions,2 and limited surgical resection may be performed for cosmetic or functionality purposes.2,3,5,6 Radiotherapy is contraindicated due to risks such as malignancy and osteonecrosis.2,3 Complications of cherubism include ocular disturbances such as proptosis and diplopia, which may rarely prolong beyond regression of the lesions of the jaw,2 and problems with speech, mastication and swallowing,2,3 which were reported by our patient.
Conclusion
Cherubism is rarely described in the African paediatric population. Histology has limited value for the diagnosis, which is primarily clinico-radiological.
Acknowledgements
The authors thank Dr Vicci du Plessis for her suggested revisions for this case report.
Competing interests
The authors declare that they have no financial or personal relationships that may have inappropriately influenced them in writing this article.
Authors' contributions
M.S.M. performed literature review and did the primary write up of the manuscript. N.N.D. assisted with the literature review and manuscript editing.
References
1. Cherubism: Best clinical practice. Orphanet J Rare Dis 2012:7(Suppl 1)S6. https://doi.org/10.1186/1750-1172-7-S1-S6 [ Links ]
2. Wagel J, Luczak K, Hendrich B, Guziński M, Sąsiadek M. Clinical and radiological features of nonfamilial cherubism: A case report. Pol J Radiol. 2012;77(3):53-57. https://doi.org/10.12659/PJR.883375 [ Links ]
3. Hille JJ, Buch B, Evans WG, Shakenovesky B, Butz S. Cherubism: Two case reports and a review of the literature. J Dent Assoc S Afr. 1986;41(7):461-466. [ Links ]
4. Beaman FD, Bancroft LW, Peterson JJ, Kransdorf MJ, Murphey MD, Menke DM. Imaging characteristics of cherubism. AJR Am J Roentgenol. 2004;182:1051-1054. https://doi.org/10.2214/ajr.182.4.1821051 [ Links ]
5. Jain V, Sharma R. Radiographic, CT and MRI features of cherubism. Pediatr Radiol. 2006;36:1099-1104. https://doi.org/10.1007/s00247-006-0261-8 [ Links ]
6. Koch BL. Cherubism. [homepage on the Internet]. Last updated 10.08.2016. [Cited 2017 Mar 28]. Available from: https://my.statdx.com/document/cherubism [ Links ]
 Correspondence:
Correspondence:
Monica Msomi
monicasheilamsomi@gmail.com
Received: 02 Nov. 2016
Accepted: 30 Jan. 2017
Published: 21 Apr. 2017

