










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSA Journal of Radiology
versão On-line ISSN 2078-6778
versão impressa ISSN 1027-202X
S. Afr. J. radiol. (Online) vol.19 no.2 Johannesburg 2015
http://dx.doi.org/10.4102/sajr.v19i2.887
CASE REPORT
Krabbe disease - An unusual presentation of optic nerve enlargement
Maria KaloianovaI; Jaishree NaidooI; Heather ThomsonII; Louisa BhenguIII
IDepartment of Radiology, Charlotte Maxeke Johannesburg Academic Hospital and University of the Witwatersrand, South Africa
IIDepartment of Paediatrics Charlotte Maxeke Johannesburg Academic Hospital and University of the Witwatersrand, South Africa
IIIDepartment of Human Genetics National Health Laboratory Services, School of Pathology and University of Witwatersrand, South Africa
ABSTRACT
Krabbe disease is an autosomal recessive leukodystrophy that presents clinically with regression of milestones, excessive irritability and inconsolable crying. The pathologic basis of the disease is abnormal myelin metabolism resulting from a deficiency in the galactocerebrosidase enzyme with subsequent white matter destruction. Although optic atrophy is a classic presentation of Krabbe disease, we report on two patients who are biological brothers presenting with optic nerve enlargement in addition to other typical magnetic resonance imaging features of Krabbe disease, thereby confounding the initial diagnosis.
Introduction
Krabbe disease, also known as globoid cell leukodystrophy, is a lysosomal function disorder that ultimately results in demyelination and dysmyelination of white matter.
The worldwide incidence of Krabbe disease has been shown to be 1 in 100 000-200 000, with 1 in 150 000 live births reported in Europe.1,2 The incidence in the South African population has not been well established.
Case 1
Patient 1 presented in February 2012 to the neurodevelopmental clinic at 7 months of age with excessive inconsolable crying, regression of milestones, numerous café au lait macules and large eyes. Birth history was non-contributory.
Magnetic resonance imaging (MRI) of the brain demonstrated hypertrophy of the optic nerves and several other cranial nerves bilaterally (Figure 1). High signal intensities were noted within the white matter of the brachium pontis, cerebellar dentate nuclei and corona radiata with a significant background of cortical and white matter atrophy (Figure 2).
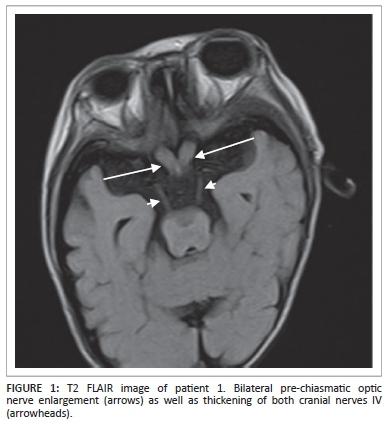
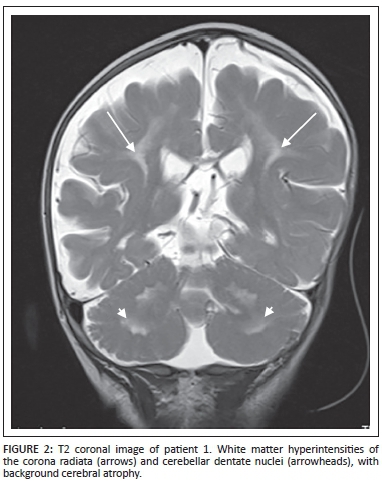
The patient died at 1 year of age with a provisional diagnosis of neurofibromatosis I (NFI). No specific tests for Krabbe disease were performed prior to his death.
Case 2
Patient 2 presented in November 2014 at 5 months of age with regression of milestones, excessive crying that was difficult to soothe, numerous café au lait macules, large eyes and a relative macrocephaly. Birth history was non-contributory and the child developed normally up until 3 months of age. Blood tests for amino acids and organic acids, as well as liver and renal functions were normal. Cerebrospinal fluid lactate and glycine levels were also normal.
Specific enzyme testing for Krabbe disease was performed for patient 2 in Philadelphia, USA. The results revealed the galactocerebrosidase enzyme value to be very low (0.07); this figure is in the range of patients affected with Krabbe disease.
MRI brain imaging of patient 2 demonstrated markedly hypertrophied optic nerves (Figure 3 and Figure 4). Increased T2 and T2 FLAIR signal intensities of the white matter of the brachium pontis and the cerebellar dentate nuclei (Figure 5) were visualised. The midbrain, pons and cerebellum also demonstrated increased T2 and T2 FLAIR signal intensities with a significant background of cortical and white matter atrophy (Figure 6).

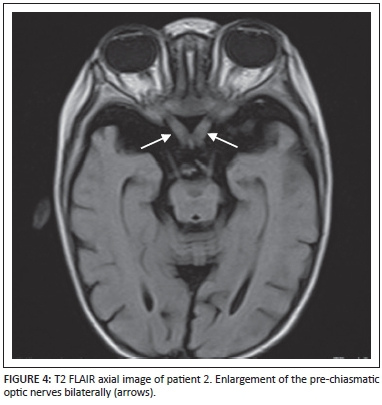


Discussion
Krabbe disease is classified as a progressive leukodystrophy with an autosomal recessive inheritance pattern. It is caused by a mutation of the galactocerebroside beta-galactosidase gene located on chromosome 14q31.2,3
Presentation is usually between 3 and 6 months of age, with death commonly occurring before the age of 2 years. The presenting features most often encountered are regression of milestones, inconsolable irritability, and agitation and feeding problems, with an inevitable need for enteric feeding in the terminal stages of the disease.2,3,4,5
The pathophysiology of Krabbe disease results from a deficiency in the production of the enzyme galactocerebrosidase, a lysosomal enzyme that forms a key component of the myelin metabolism pathway.3 This deficiency results in progressive demyelination and dysmyelination of both the peripheral and central nervous systems.4,6
The impaired galactocerebrosidase production results in accumulation of lethal levels of galactocerebroside and psychosine within macrophages, with resultant globoid cell formation.4,7 Psychosine in particular has been shown to be toxic to oligodendroglial cells, neurons and Schwann cells.1,3
The microscopic features of Krabbe disease typically include severe neuronal destruction in the white matter of the thalami, cerebellar peduncles, corona radiata and corpus callosum.3
Computed tomography (CT) usually shows hyperdensities in the thalami, caudate nuclei, cerebellar dentate nuclei and corona radiata during the early stages of the disease.3 As the disease progresses, associated white matter atrophy manifests, making it indistinguishable from other dysmyelinating conditions in the terminal stage.3,6
In very young patients, T1-weighted MRI demonstrates abnormally high signal in the thalami, with a delay in the appearance of the normal high signal in the internal capsules. High T1 signal intensities are also observed in the caudate nuclei, corona radiata, dentate nuclei of the cerebellum and also within the cerebral and cerebellar white matter. In the early stages of the disease, the subcortical white matter is spared.3,6
The cerebellar dentate nuclei maintain their high signal on T2-weighted imaging. In addition, there are also abnormally high T2 signal intensities within the corticospinal tracts of the internal capsules, cerebral white matter and the medial medullary pyramids. These findings are highly indicative of the disease.3
Beyond the first year of the disease, white matter high T2 signal intensities are noted to involve the splenium of the corpus callosum and the deep cerebral white matter, notably in the posterior frontal and anterior parietal lobes.3
Diffusion weighted imaging (DWI) demonstrates proportionally increased diffusion with disease progression, noted uniformly throughout the white matter. MR spectroscopy, although non-specific, may show an increased lactate and decreased N-acetylaspartate (NAA).3
Optic nerve as well as other cranial nerve enlargement, however, is seldom described in association with Krabbe disease. When present, it is thought to be as a result of globoid cell accumulation within the optic and other cranial nerves in combination with an inflammatory response related to myelin breakdown.3,4,5,6,8 Jones et al. proved the presence of globoid cells within the optic nerves on histological review of one of their analysed cases.6
In the presence of optic nerve enlargement, the differential diagnosis will include optic nerve glioma in patients with NF-I, optic dural ectasia, granulomatous or histiocytic infiltration of the optic nerves, post-viral optic neuritis and optic nerve medulloepithelioma.6,7 Making the distinction on clinical grounds is of great importance.
Conclusion
Optic atrophy is one of the clinical presenting features of Krabbe disease. However, in rare cases as we report here, optic nerve enlargement may be an association. Considering the higher prevalence of NF-I, one can easily be misled by these imaging features into making the latter diagnosis.
Early awareness of neuroimaging findings in Krabbe disease together with a suggestive clinical presentation will allow timely management and genetic counseling to parents.
Acknowledgements
We thank Professor L.B. Jacklin and Dr J. Epstein of the Neurodevelopmental Unit, Department of Paediatrics, Charlotte Maxeke Johannesburg Academic Hospital and University of the Witwatersrand, Johannesburg, who were the clinicians involved in the care of the above patients. In addition, we thank Dr D.A. Wenger, Director, Lysosomal Diseases Testing Laboratory, Sidney Kimmel Medical College, Thomas Jefferson University, Philadelphia, USA, for assisting us in confirming the diagnosis.
Competing interests
The authors declare that they have no financial or personal relationships which may have inappropriately influenced them in writing this article.
Authors' contributions
M.K. (University of the Witwatersrand) and J.N. (University of the Witwatersrand) contributed towards the imaging and literature review. H.T. (University of the Witwatersrand) and L.B. (University of the Witwatersrand) contributed towards the clinical information and confirmation of the diagnosis.
References
1. Nagar VA, Ursekar MA, Krishnan P, Jankharia BG. Krabbe disease: Unusual findings. Pediatr Radiol. 2006;36:61-64. PMID: 16247616. http://dx.doi.org/10.1007/s00247-005-0008-y [ Links ]
2. Korn-Lubetzki I, Dor-Wollman T, Soffer D, Raas-Rothschild A, Hurvitz H, Nevo Y. Early peripheral nervous system manifestations of infantile Krabbe disease. Pediatr Neurol. 2003;28:115-118. PMID: 12699861, http://dx.doi.org/10.1016/S0887-8994(02)00489-7 [ Links ]
3. Barkovich AJ, Raybaud C. Pediatric neuroimaging. 5th edn. Philadelphia: Lippincott Williams and Wilkins, 2012; p. 92-93. [ Links ]
4. Beslow LA, Schwartz ES, Bönnemann CG. Thickening and enhancement of multiple cranial nerves in conjunction with cystic white matter lesions in early infantile Krabbe disease. Pediatr Radiol. 2008;38:694-696. PMID: 18265968, http://dx.doi.org/10.1007/s00247-008-0763-7 [ Links ]
5. Patel B, Gimi B, Vachha B, Agadi S, Koral K. Optic nerve and chiasm enlargement in a case of infantile Krabbe disease: Quantitative comparison with 26 age-matched controls. Pediatr Radiol. 2008;38:697-699. PMID: 18438656, http://dx.doi.org/10.1007/s00247-008-0849-2 [ Links ]
6. Jones BV, Barron TF, Towfighi J. Optic nerve enlargement in Krabbe's disease. AJNR Am J Neuroradiol. 1999;20:1228-1231. PMID: 10472976. [ Links ]
7. Hussain SA, Zimmerman HH, Abdul-Rahman OA, Hussaini SM, Parker CC, Khan M. Optic nerve enlargement in Krabbe disease: A pathophysiologic and clinical perspective. J Child Neurol. 2011;26:642-664. PMID: 21285037, http://dx.doi.org/10.1177/0883073810387929 [ Links ]
8. Shah S, Freeman E, Wolf V, Murthy S, Lotze T. Teaching neuroimages: Intracranial optic nerve enlargement in infantile Krabbe disease. Neurology. 2012;78:e126. PMID: 22585439, http://dx.doi.org/10.1212/WNL.0b013e3182563bad [ Links ]
 Correspondence:
Correspondence:
Maria Kaloianova
PO Box 785085, Sandton 2146
South Africa
mkaloianova@gmail.com
Received: 07 June 2015
Accepted: 14 Aug. 2015
Published: 30 Oct. 2015

