










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSA Journal of Radiology
On-line version ISSN 2078-6778
Print version ISSN 1027-202X
S. Afr. J. radiol. (Online) vol.19 n.1 Johannesburg 2015
http://dx.doi.org/10.4102/sajr.v19i1.929
QUIZ CASE
Answer to quiz case: Temporal bone imaging
Enrico ArkinkI, II; J. FrijnsIII; Berit VerbistI, IV
IDepartment of Radiology, Leiden University Medical Center, The Netherlands
IIDepartment of Radiology, Medical Center Haaglanden, The Netherlands
IIIDepartment of Otorhinolaryngology, Leiden University Medical Center, The Netherlands
IVDepartment of Radiology, Radboud University Nijmegen Medical Center, The Netherlands
ABSTRACT
Computed tomographic scanning of the petrous bone and magnetic resonance imaging sequences of the inner ear and cerebellopontine angle of a deaf patient were performed to find an explanation for his deafness, and to establish whether he would be a good candidate for cochlear implantation. The imaging features were considered pathognomonic for incomplete partition type III (IP type III). Further management and discussion of this deafness subtype are detailed.
Introduction
The best responses received for this temporal bone imaging quiz case were from Dr T.E. Buthelezi and Dr Herman van Vuuren. Congratulations to them.
Background
Computed tomographic (CT) scanning of the petrous bone and magnetic resonance imaging (MRI) sequences of the inner ear and cerebellopontine angle of a deaf patient were performed to find an explanation for his deafness, and to establish whether he would be a good candidate for cochlear implantation.
Diagnosis
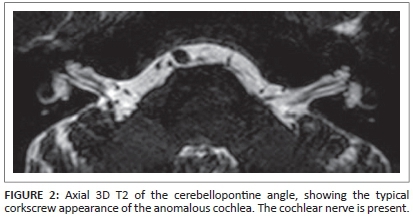
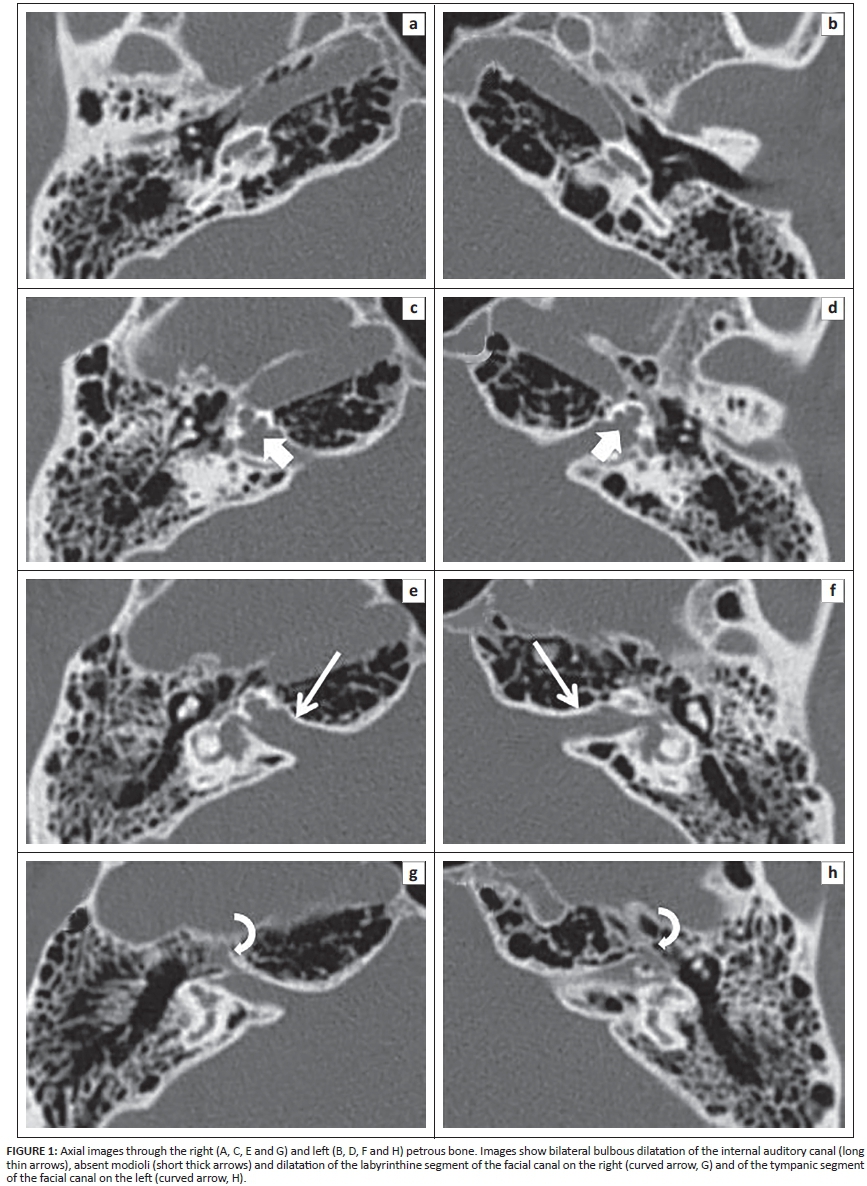
The CT images of the temporal bone showed a normal aspect of the mastoid, tympanic membrane middle ear cavity and ossicular chain bilaterally (not shown in detail in Figure 1). The abnormalities can be seen in the inner ear structures (Figure 1). The images show bilateral bulbous dilatation of the internal auditory canal (IAC). The bone plates at the fundus of the IAC, which separate them from the cochlear basal turn, are absent. The modiolus is deficient, with a present interscalar septum, giving the dysplastic cochleas a corkscrew appearance. This corkscrew appearance is also apparent on 3D T2 MR-imaging (Figure 2). Also visible is the minor dilatation of the labyrinthine segment of the facial canal on the right, and of the tympanic segment of the facial canal on the left (Figure 1). These specific imaging features are considered pathognomonic for incomplete partition type III (IP type III).
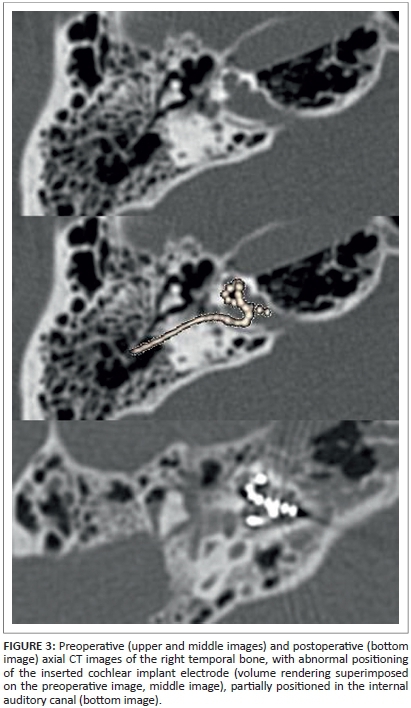
The patient underwent cochlear implantation on the right side. Insertion of the electrode of the cochlear implant was troublesome but complete. Upon cochleostomy, expected perilymph gushing owing to a fistulous connection between the IAC and the cochlea occurred. The gushing was controlled and properly sealed. Perioperative impedance measurements of the electrodes and brainstem responses showed good measurements for apical and basal electrode contacts, but not for those in between. Postoperative imaging confirmed an unusual position of the electrode, with parts of it bulging into the IAC (Figure 3). Apparently, the tip of the electrode had been blocked in the upper basal turn during insertion, causing part of the middle portion of the array to bulge into the internal auditory meatus, when the surgeon tried to achieve full insertion. Despite this unfavourable electrode position, the patient recovered well from surgery and, with an adequate fitting, postoperative performance of this prelingually deaf adult was acceptable (up to 55% of phonemes correct on a monosyllabic word test on a CD in quiet), with the patient able to hear sufficiently to participate normally in conversation.
Discussion
The combination of clinical and imaging findings in this patient are characteristic of IP type-III, which is often also referred to as X-linked deafness type 2 (DFNX2).1,2,3,4,5,6,7,8,9 This inner ear anomaly is a rare congenital cause of progressive mixed (conductive and sensorineural) hearing loss. It is the rarest form of incomplete partitioning, representing about 2% of all inner ear malformations.10
The simultaneous occurrence of bilateral bulbous dilatation of the IAC, absence of the lamina cribrosa separating the IAC from the cochlear basal turn, absent modioli and dilatation of the first two segments of the facial canal should make one aware of the diagnosis of IP type-III. Glasscock, using coronal section polytomography,11 recognised the dilatation of the IAC already in the early 1970s. With improving technology, more detailed descriptions were published,12,13 with dilatation of the facial canal noted for the first time in the 1980s.14 In a few DFNX2 patients, medialisation of the origin of the vestibular aqueduct and slight dilatation of this structure have also been described.5 The modiolus is completely absent, but interscalar septa are present.15 The cochlea is positioned at the lateral end of the IAC instead of the usual anterolateral position, resulting in the corkscrew appearance10 but, contradictory to the initial reports using polytomography,4 is considered to have normal external diameters.16 The slight dilatation of the first two segments of the intratemporal facial nerve may be accompanied by an abnormal, superior position of the labyrinthine segment in relation to the cochlea. In addition, enlargement of superior vestibular nerve canals has been reported in some patients with IP type-III.9 The round window may be small and artretic.9
The absence of the bony partition between the IAC and cochlea allows a direct fistulous connection between the subarachnoid space and the perilymph in the labyrinth. Intracranial pressure is transmitted to the perilymphatic spaces (scala vestibuli and scala tympani), leading to an increased pressure impinging on the cochlear duct17; this may cause progressive inner ear dysfunction, eventually leading to sensorineural hearing loss. Patients may also show impaired vestibular function, as demonstrated in the current patient.18 DFNX2 is also associated with fixation of the (thickened) stapedial footplate, leading to the alternative term 'conductive deafness with stapes fixation' (DFN3). The increased pressure in the perilymphatic compartment, in particular within the vestibule, interferes with normal movement of the stapedial footplate, resulting in immobilisation.17 In this respect, the audiogram can be misleading, and suggest a false diagnosis of otosclerosis. Stapedectomy, in an attempt to improve the conductive portion of the hearing loss, is, contraindicated in affected individuals, however. Removal of the immobilised stapedial footplate results in a perilymphatic gusher owing to the fistulous connection between the internal auditory canal and the cochlea.19 Because of attempted stapedectomy with resulting leakage in the past, the anomaly is sometimes referred to as 'X-linked stapes gusher'.
DFNX2 is caused by a mutation in or around the POU3F4 gene on chromosome Xq21.20 It is one of at least five loci implicated in congenital deafness.21,22 It is found in approximately 50% of families with X-linked hearing loss.23 Most of the other types of X-linked hearing loss, such as X-linked deafness type 3 (DFNX3, DFN4), show no evidence of radiographic abnormalities of the temporal bone. However, the X-linked congenital deafness that was ascribed to a mutation on the type IV collagen gene COL4A6 recently showed CT findings remarkably similar to DFNX2,21 which questions whether the terms IP type III and DFNX2 should be used interchangeably. Because of the X-linked recessive inheritance pattern, affected individuals with this type of mixed hearing loss are mostly male. These male patients typically present with profound hearing loss at birth, rapidly progressing to severe deafness within the first decade of life.23 Although similar CT findings have been observed in affected female patients,24 female carriers tend to have milder forms of the same anomaly,4,18 resulting in normal hearing or only mild, moderate or delayed-onset hearing loss.1,9 However, CT imaging should be obtained in female patients with suspected family history who present with clinical features of the disease.24 Despite the absence of a family history, as in our patient, a radiologist should suggest the diagnosis of DFNX2 when typical inner ear abnormalities are seen on CT. MRI with a 3D constructive interference in steady state (CISS) should be obtained to confirm the presence of cochleovestibular nerves in the event that cochlear implantation is considered. Preferably, an experienced neuroradiologist should interpret the imaging studies.9 A correct imaging diagnosis before surgical intervention might avoid a perilymphatic gusher and consequent complications, such as increased hearing loss or (recurrent) meningitis, in the patient, or even in other family members with hearing loss. Recurrent meningitis may also occur spontaneously.8 In the patient presented here, neither genetic testing nor screening of relatives was performed.
With normal outer diameters, but disorganised internal architecture of the cochlea, IP type III can be treated successfully with cochlear implantation when sensorineural hearing loss is profound.9 Apart from regular complications of cochlear implants, such as facial nerve stimulation or device failure, two major problems may occur peroperatively in IP type III: perilymphatic gusher when the cochlea is opened (cochleostomy), and misplacement of the cochlear implant electrode in the IAC. Because of this possibility of electrode malpositioning, cochlear implantation in IP type III was initially not advised.25 To prevent electrode misplacement, the surgeon should carefully consider the type of electrode to be used.9,10
In summary: IP type III is a rare cause of mixed conductive and sensorineural hearing loss, but the radiologist may play a crucial role in both diagnosis and treatment of the disorder.
Competing interests
The authors declare that they have no financial or personal relationships which may have inappropriately influenced them in writing this article.
Authors' contributions
E.B.A. (Leiden University Medical Center; Medical Center Haaglanden) drafted the manuscript. J.F. (Leiden University Medical Center) and B.V. (Leiden University Medical Center; Radboud University Nijmegen Medical Center) critically revised it for important intellectual content.
References
1.Altay H, Savas R, Ogut F, Kirazli T, Alper H. CT and MRI findings in X-linked progressive deafness. Diagn Interv Radiol. 2008;14:117-119. PMID: 18814129. [ Links ]
2. Gong WX, Gong RZ, Zhao B. HRCT and MRI findings in X-linked non-syndromic deafness patients with a POU3F4 mutation. Int J Pediatr Otorhinolaryngol. 2014;78: 1756-1762. PMID: 25175280, http://dx.doi.org/10.1016/j.ijporl.2014.08.013 [ Links ]
3. Kumar G, Castillo M, Buchman CA. X-linked stapes gusher: CT findings in one patient. AJNR Am J Neuroradiol. 2003;24:1130-1132. PMID: 12812938. [ Links ]
4. Phelps PD, Reardon W, Pembrey M, Bellman S, Luxom L. X-linked deafness, stapes gushers and a distinctive defect of the inner ear. Neuroradiology. 1991;33:326-330. PMID: 1922747, http://dx.doi.org/10.1007/BF00587816 [ Links ]
5. Talbot JM, Wilson DF. Computed tomographic diagnosis of X-linked congenital mixed deafness, fixation of the stapedial footplate, and perilymphatic gusher. Am J Otol. 1994;15:177-182. PMID: 8172298. [ Links ]
6. Tang A, Parnes LS. X-linked progressive mixed hearing loss: Computed tomography findings. Ann Otol Rhinol Laryngol. 1994;103:655-657. PMID: 8060062, http://dx.doi.org/10.1177/000348949410300814 [ Links ]
7. Chee NW, Suhailee S, Goh J. Clinics in diagnostic imaging (111): X-linked congenital mixed deafness syndrome. Singapore Med J. 2006;47:822-824. PMID: 16924369. [ Links ]
8. Alizadeh H, Nasri F, Mehdizadeh M, Jamsa S. Computed tomography findings of a patient with severe dysplasia of the inner ear and recurrent meningitis: A case report of gusher ear in a five-year old boy. Iran J Radiol. 2014;11:e4168. PMID: 25763081, http://dx.doi.org/10.5812/iranjradiol.4168 [ Links ]
9. Incesulu A, Adapinar B, Kecik C. Cochlear implantation in cases with incomplete partition type III (X-linked anomaly). Eur Arch Otorhinolaryngol. 2008;265:1425-1430. PMID: 18305951, http://dx.doi.org/10.1007/s00405-008-0614-z [ Links ]
10. Naito Y. Pediatric ear diseases. Basel: Karger; 2013. [ Links ]
11. Glasscock ME, III. The stapes gusher. Arch Otolaryngol. 1973;98:82-91. PMID: 4723769, http://dx.doi.org/10.1001/archotol.1973.00780020088004 [ Links ]
12. Bento RF, Miniti A. X-linked mixed hearing loss: Four case studies. Laryngoscope. 1985;95:462-468. PMID: 4039021, http://dx.doi.org/10.1288/00005537-198504000-00017 [ Links ]
13. Jensen J, Terkildsen K, Thomsen KA. Inner ear malformations with oto-liquorrhea. Tomographic findings in three cases with a mixed hearing impairment. Arch Otorhinolaryngol. 1977;214:271-282. PMID: 576408, http://dx.doi.org/10.1007/BF00458322 [ Links ]
14. Cremers CW, Hombergen GC, Wentges RT. Perilymphatic gusher and stapes surgery. A predictable complication? Clin Otolaryngol Allied Sci. 1983;8:235-240. PMID: 6652936, http://dx.doi.org/10.1111/j.1365-2273.1983.tb01434.x [ Links ]
15. Sennaroglu L. Cochlear implantation in inner ear malformations-A review article. Cochlear Implants Int. 2010;11:4-41. PMID: 19358145, http://dx.doi.org/10.1002/cii.416 [ Links ]
16.Sennaroglu L, Sarac S, Ergin T. Surgical results of cochlear implantation in malformed cochlea. Otol Neurotol. 2006;27:615-623. PMID: 16788416, http://dx.doi.org/10.1097/01.mao.0000224090.94882.b4 [ Links ]
17. Swartz JD, Loevner LA. Imaging of the temporal bone. New York: Thieme; 2009. [ Links ]
18. Van De Water T, Staecker H. Otolaryngology: Basic science and review. New York: Thieme; 2006. [ Links ]
19. Nance WE, Setleff R, McLeod A, Sweeney A, Cooper C, McConnell F. X-linked mixed deafness with congenital fixation of the stapedial footplate and perilymphatic gusher. Birth Defects Orig Artic Ser. 1971;7:64-69. PMID: 5173351. [ Links ]
20. de Kok YJ, van der Maarel SM, Bitner-Glindzicz M, et al. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science. 1995;267:685-688. PMID: 7839145, http://dx.doi.org/10.1126/science.7839145 [ Links ]
21. Rost S, Bach E, Neuner C, et al. Novel form of X-linked nonsyndromic hearing loss with cochlear malformation caused by a mutation in the type IV collagen gene COL4A6. Eur J Hum Genet. 2014;22:208-215. PMID: 23714752, http://dx.doi.org/10.1038/ejhg.2013.108 [ Links ]
22. Song MH, Lee KY, Choi JY, Bok J, Kim UK. Nonsyndromic X-linked hearing loss. Front Biosci (Elite Ed). 2012;4:924-933. PMID: 22201925. [ Links ]
23. Petersen MB, Wang Q, Willems PJ. Sex-linked deafness. Clin Genet. 2008;73: 14-23. PMID: 18005182. [ Links ]
24. Papadaki E, Prassopoulos P, Bizakis J, Karampekios S, Papadakis H, Gourtsoyiannis N. X-linked deafness with stapes gusher in females. Eur J Radiol. 1998;29:71-75. PMID: 9934561, http://dx.doi.org/10.1016/S0720-048X(98)00027-8 [ Links ]
25. Phelps PD. Cochlear implants for congenital deformities. J Laryngol Otol. 1992;106: 967-970. PMID: 1479272, http://dx.doi.org/10.1017/S0022215100121486 [ Links ]
 Correspondence:
Correspondence:
Enrico Arkink
PO Box 9600, 2300 RC Leiden
The Netherlands
Email: e.b.arkink@lumc.nl
Received: 26 Aug. 2015
Accepted: 28 Aug. 2015
Published: 21 Oct. 2015


{kind=link}