










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSA Journal of Radiology
versão On-line ISSN 2078-6778
versão impressa ISSN 1027-202X
S. Afr. J. radiol. (Online) vol.19 no.1 Johannesburg 2015
http://dx.doi.org/10.4102/SAJR.v19i1.792
QUIZ CASE
Answer to musculoskeletal imaging quiz case
Shalendra Misser; John Zietkiewicz
Lake Smit and Partners Inc., Durban, South Africa
Case findings and diagnosis
No correct answer was received for this musculoskeletal quiz case.
The patient, a 34-year-old female runner, presented with a 6-week history of right hip pain that progressed slowly. She curtailed her physical activity, but was still able to continue with her other daily living activities. At the time of initial presentation, a plain radiograph (Figure 1) was performed, demonstrating subtle lytic foci in the right supra-acetabular ileum and right lesser trochanter. No surrounding sclerotic rim or soft-tissue fat plane displacement was noted. Preliminary radiographic differential diagnoses included multiple myeloma, osteolytic metastatic disease, eosinophilic granulomas or cystic angiomatosis. Initial blood tests confirmed normal haemoglobin, serum calcium, liver function, urea and electrolytes.

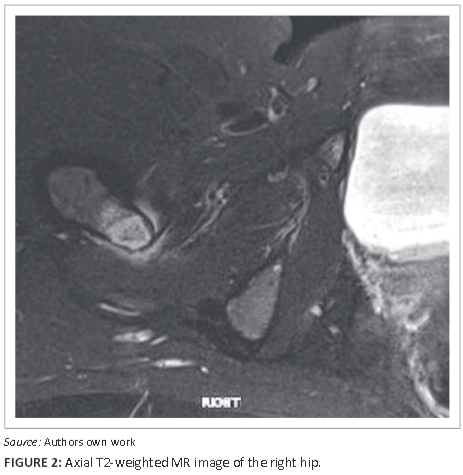
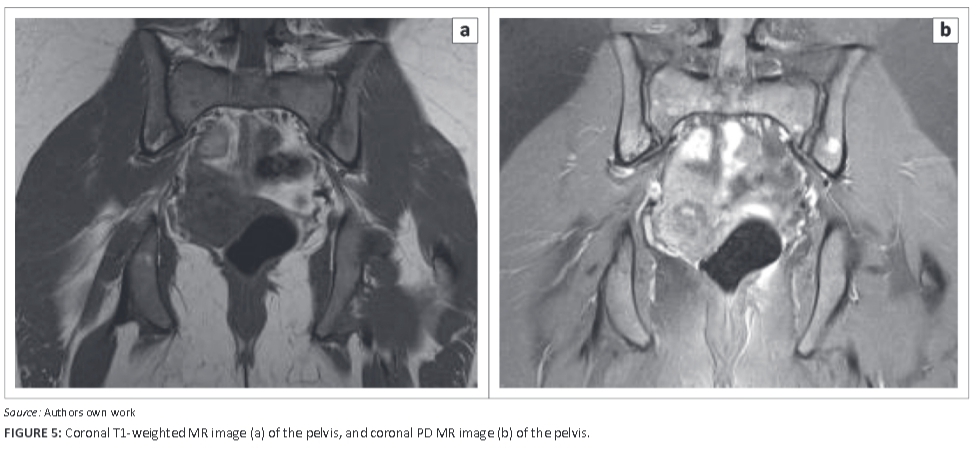
Figure 2 and Figure 3 are axial T2-weighted and sagittal proton-density (PD)-weighted magnetic resonance images respectively of the right hip, showing the circumscribed hyperintense lesser trochanteric lesion with reactive surrounding soft-tissue hyperintensity including the iliopsoas tendon insertion. A computed tomography (CT) scan of the chest (Figure 4) suggested borderline splenic enlargement with areas of low attenuation in the body of the spleen. The other organs were not involved. Figure 5a and Figure 5b are coronal T1 and PD images of the pelvic girdle demonstrating several focal lesions, with T1 shortening and corresponding hyperintensity on the PD sequence. The lesions, of varying size, measured up to a centimeter, with preserved margins, no significant periosteal reactions or adjacent soft-tissue masses.


A CT scan of the pelvis (Figure 6a) was performed on follow-up to re-evaluate the lesser trochanter focus. This showed a little internal density within the trochanteric lesion and small lytic foci at both iliac bones. CT angiography (Figure 6b) did not show any significant neovascularisation or prominent vasculature of aneurysmal bone cysts. We performed a CT-guided biopsy (Figure 7) of the right lesser trochanteric lesion. Histopathological analysis demonstrated cystic vascular spaces, lined by a single layer of endothelial cells, separated by thin intervening spindled stroma. Immunochemistry showed CD34/CD31 positivity, and the histological conclusion was that of a vasoformative lesion with local bony destruction compatible with cystic angiomatosis.

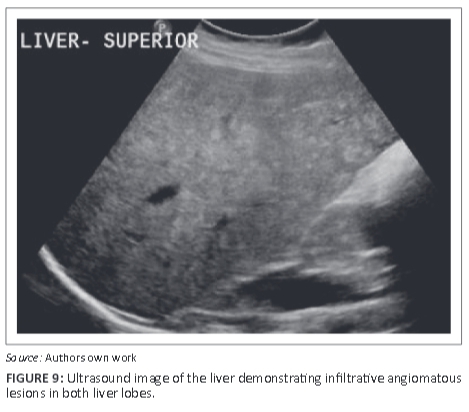
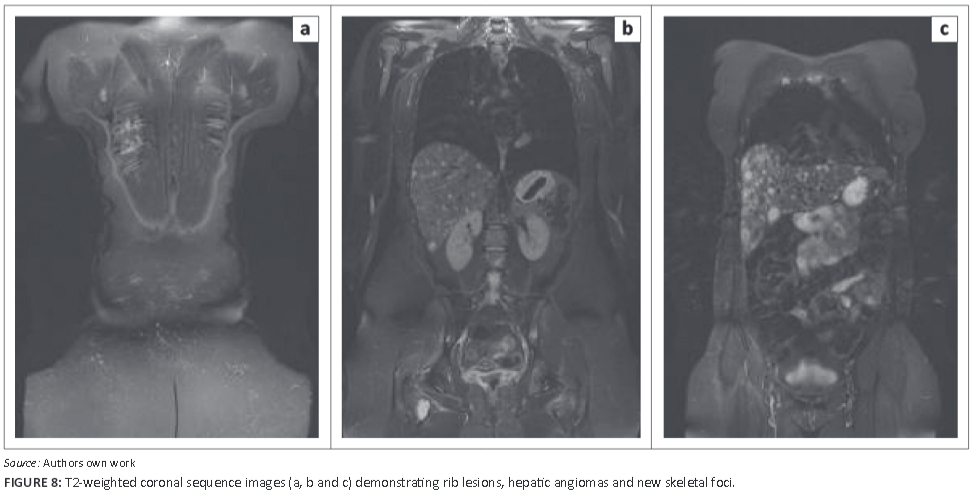
As the patient had single visceral organ involvement, splenectomy was subsequently performed. She has since had close clinical and radiological follow-up. Unfortunately, at recent follow-up the patient was found to have several angiomatous lesions of her liver and several further skeletal lesions, and she is currently unwell. Figure 8a to Figure 8c are selected images from recent magnetic resonance scanning - whole body screening, T2-weighted sequence. Figure 8a demonstrates several rib lesions. Figure 8b shows the presence of angiomas of the liver, and new bony lesions at the humeri and in the spine. Figure 8c reveals the more confluent angiomatous infiltration of the right lobe of the liver anteriorly. Ultrasound of the liver (Figure 9) shows the infiltrative mixed, predominantly echogenic, lesions of the liver in keeping with multiple angiomas. Her prognosis is now poor.
Discussion
Cystic angiomatosis of bone (CAB) is a rare pathologic entity characterised by several cysts of varying sizes involving the skeleton. First described in 1953 by Jacobs and Kimmelsteil,1 several synonyms have been used including haemangiomatosis, lymphangiomatosis and hamartomatous haemolymphangiomatosis. The reason for this overlap between haemangioma and lymphangioma is most apparent on histopathology where, even in an individual patient, there may be significant variability in the degree of expression. Some areas of a cystic lesion may be filled with blood constituents whilst others may contain proteinaceous material akin to haemangiomas and lymphangiomas respectively.2,3,4,5
CAB is usually a disease of younger patients affecting adolescents and young adults, and rarely occurring in older persons. There is no significant gender predilection; however, some authors indicate a slightly increased incidence in young men.3,4 Patients with CAB may be completely asymptomatic or may experience some bone pain even in the absence of any pathological fractures.2
The clinical course and severity depends largely on the presence of visceral and soft-tissue involvement, which has been reported in up to 60%-70% of affected individuals.2 The spleen is almost always involved in such instances. Other organs that may be involved include the pleura, lung, peritoneum, liver, kidneys, mesentery and lymphatic tissue. In these patients, CAB has been referred to as 'skeletal-extraskeletal angiomatosis',6 with the disease more progressive, and it can result in early death.5
Concepts regarding the aetiology of CAB have varied in the last six decades. In 1972, Boyle3 purported two possible pathomechanisms: (1) a true metastasising angioma or (2) a multicentric congenital malformation such as a vascular hamartoma. Support has followed for the latter in more recent times.4
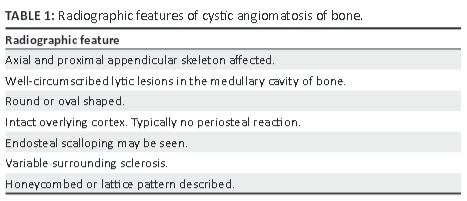
CAB commonly affects the axial skeleton, particularly the pelvis, ribs, spine and skull. Of the appendicular skeleton, the femur, humerus and scapula may also be frequently involved, but hands and feet are usually spared. Roentgen features of CAB (Table 1) include well-circumscribed round or ovoid cysts, usually numerous, with some lesions that may show expansion and peripheral sclerosis. The sparing of the overlying cortex and absence of periosteal reactions are typical, but the converse may be true if a pathological fracture develops. A honeycomb or lattice-work pattern is also described, and resembles a solitary bony haemangioma radiographically.5
On CT scans, lesions are lytic and well-circumscribed, sometimes demonstrating peripheral sclerosis. Pathological fractures are better delineated on CT scanning, and multiplicity, which may be radiographically occult, becomes more obvious. MRI signal characteristics of CAB lesions include T1-weighted low-to-intermediate signal intensity depending on the internal fat content within the lesion. Corresponding T2-weighted high-signal intensity is shown in CAB lesions owing to vascular and cystic/fluid components.2
There is a wide differential diagnosis (Table 2) for CAB. Eosinophilic granulomas typically occur in younger patients, before age 30, and can affect the skull up to five times more frequently than any other site.3Hepatosplenomegaly is a feature of Letterer-Siwe disease but this syndrome, associated with histiocytosis, largely occurs in very young children. Furthermore, cortical destruction with coalescence of adjacent lesions is commonly seen in eosinophilic granulomas.
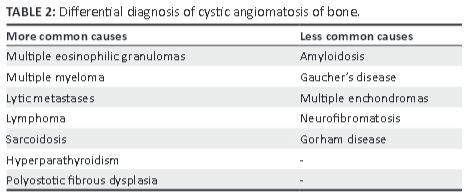
Multiple myeloma is rare in persons under the age of 40 years. Lytic lesions tend to be of similar size and, when present, relate to earlier disease progression. Serum protein electrophoresis can identify a gammopathy, aiding diagnosis confirmation. Sarcoidosis can affect bone with non-caseating granulomas resulting in solitary or multiple lytic lesions that may be expansile and tend to involve bony cortex in addition to the medullary cavity. Visceral involvement in sarcoidosis and storage diseases such as Gaucher's disease may lead to hepatosplenomegaly, making these mimickers of CAB. One should look for additional features of Erlenmeyer flask deformities of the femora or aseptic necrosis in patients with Gaucher's disease.
In hyperparathyroidism, the cystic bone lesions generally lack a surrounding rim of sclerosis, which may also be seen in CAB. Skull involvement is rare, and vertebral involvement owing to renal osteodystrophy may produce a 'rugger jersey' spine appearance. Polyostotic fibrous dysplasia lesions are usually unilateral, and the bony lesions demonstrate an opalescent ground glass appearance. A skull base predilection is common and, conversely, visceral involvement is unusual in fibrous dysplasia. Enchondromas are more commonly solitary and generally demonstrate intralesional calcification with matrix mineralisation, resulting in some internal T2-weighted low signal. Gorham disease, which may be histologically indistinguishable from CAB, is particularly destructive, with solitary osseous involvement being the rule.
Treatment for CAB is largely symptomatic. Analgesics suffice for bone pain and, when bony remodelling occurs, biphosphonates may be prescribed. Occasionally, surgery is required for bony lesions including for the repair of displaced pathological fractures. Local radiotherapy and chemotherapy have been used for skeletal lesions but these measures have not been shown to alter the outcome. If the spleen is the sole visceral organ involved, splenectomy may be performed.2,6
CAB has radiographic features that are not specific and overlap considerably with other vascular or lytic lesions of bone. A radiologist's knowledge of this entity and its imaging characteristics is vital in guiding the clinical investigation and management of affected individuals.
Acknowledgements
Competing interests
The authors declare that they have no financial or personal relationships which may have inappropriately influenced them in writing this article.
References
1.Jacobs JF, Kimmelstiel P. Cystic angiomatosis skeletal system. J Bone Joint Surg Am. 1953;35-A:409-420. [ Links ]
2. Ballina-Garcia FJ, Queiro-Silva MR, Molina-Suarez R, Fernandez-Martinez J, Rivela-Vasquez M, Rodriguez-Perez A. Multiple painful bone cysts in a young man. Ann Rheum Dis. 1996;55:346-349.http://dx.doi.org/10.1136/ard.55.6.346 [ Links ]
3. Boyle WJ. Cystic angiomatosis of bone. A report of three cases and review of the literature. J Bone Joint Surg Br. 1972;54:626-636. [ Links ]
4. Lateur L, Simeons CJ, Gryspeerdt S, Samson I, Mertens V, Van Damme B. Skeletal cystic angiomatosis. Skeletal Radiol. 1996;25:92-95. http://dx.doi.org/10.1007/s002560050042 [ Links ]
5. Murphey MD, Fairbairn KJ, Parman LM, Baxter KG, Parsa MB, Smith WS. From the Archives of the AFIP. Musculoskeletal angiomatous lesions: Radiologic-pathologic correlation. Radiographics. 1995;15:893-917. http://dx.doi.org/10.1148/radiographics.15.4.7569134 [ Links ]
6. Clayer M. Skeletal angiomatosis in association with gastro-intestinal angiodysplasia and paraproteinemia: A case report. J Orthop Surg (Hong Kong). 2002;10:85-88. [ Links ]
 Correspondence:
Correspondence:
Shalendra Misser
Unit 301, 7 Poynton Place
Musgrave 4001, Durban, South Africa
Email: misser@lakesmit.co.za
Received: 13 Feb. 2015
Accepted: 18 Feb. 2015
Published: 04 June 2015


{kind=link}
{kind=link}
{kind=link}