










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSA Journal of Radiology
versão On-line ISSN 2078-6778
versão impressa ISSN 1027-202X
S. Afr. J. radiol. (Online) vol.19 no.1 Johannesburg 2015
http://dx.doi.org/10.4102/SAJR.v19i1.760
PICTORIAL INTERLUDE
Double trouble: Bilateral cerebral involvement in Sturge-Weber syndrome
Narosha Adroos; Janet Smal; Farhana E. Suleman
Department of Radiology, University of Pretoria, South Africa
ABSTRACT
Sturge-Weber syndrome, also known as encephalotrigeminal angiomatosis or meningofacial angiomatosis, is characterised in its classical form by a congenital, usually unilateral, 'port-wine stain' (capillary naevus) on the face, convulsions, typical intracranial calcification and some degree of mental retardation and hemiparesis. The clinical correlation of intractable seizures with the presence of bilateral intracranial disease has management and prognostic implications, thus making the presence of bilateral disease an important factor to all those involved in the management of the child with Sturge-Weber syndrome.
Introduction
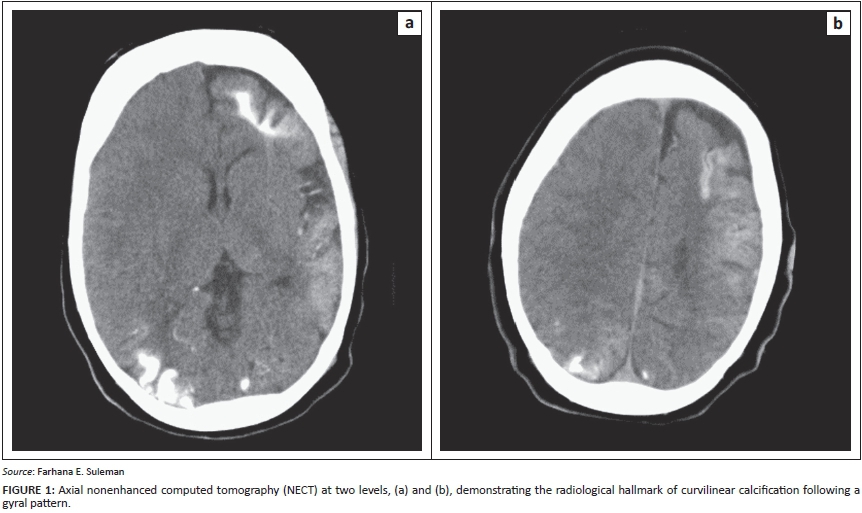
Sturge-Weber syndrome (SWS), an uncommon condition that forms part of the group of phakomatoses, is also known as encephalotrigeminal angiomatosis or meningofacial angiomatosis. It is characterised by a congenital, usually unilateral, 'port-wine stain' (capillary naevus) affecting the face, convulsions, typical intracranial gyral calcification and tram tracking (Figure 1), some degree of mental retardation and hemiparesis.1,2 It is a progressive disorder, ultimately associated with profound neurological decline, the mechanism of which is incompletely understood.
Bilateral disease may occur in up to 15% of patients diagnosed with SWS; however, this probability is not mentioned in any of the widely used radiology, neurology or paediatrics textbooks.2
Discussion
The aetiology of SWS is thought to be the result of residual embryonal blood vessels and their secondary effects on surrounding brain tissue. A vascular plexus develops around the cephalic portion of the neural tube, under ectoderm destined to become facial skin. Normally, this vascular plexus forms in the sixth week and regresses around the ninth week of gestation. Failure of this normal regression results in residual vascular tissue, which forms the angiomata of the leptomeninges, face and ipsilateral eye. Neurological dysfunction results from secondary effects on surrounding brain tissue, which include hypoxia, venous occlusion, thrombosis, infarction, or vasomotor phenomenon.1
Patients with SWS are particularly susceptible to brain injury as a consequence of status epilepticus and require optimal seizure control. Bilateral cutaneous involvement is seen in 32.5% of cases as compared to bilateral intra-cranial involvement seen in only 7.5% of cases. Patients with bilateral intracranial disease are usually more severely affected with intractable seizures; therefore the diagnosis of unexpected bilateral SWS is clinically important as urther ischaemic injury reduces the cortex to a calcified non-functioning mantle.3
Diagnosis of SWS is usually made on clinical findings, and imaging is useful for confirming the diagnosis. In severe cases, the seizures become resistant to medical treatment and surgery may be considered, thus making diagnostic imaging essential to define the full extent of the disease.
According to Hoffman et al.,4 patients with seizures associated with SWS who do not respond to medication can be candidates for surgery in the form of hemispherectomy to remove the seizure focus or less radical lobar surgery. Sectioning of the corpus callosum has been described in an attempt to prevent extension of seizures into the normal hemisphere from the abnormal side.5 In such cases, imaging is very important, firstly to exclude or confirm the presence of bilateral SWS, and secondly to determine the full extent of leptomeningeal and cortical disease that would facilitate less radical resection.6
Imaging findings
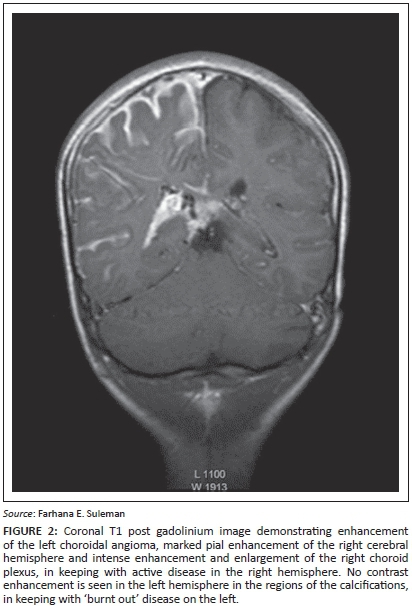
Magnetic resonance imaging (MRI) post contrast enhancement is the most accurate imaging study to demonstrate the extent of the pial enhancement, as cortical calcifications usually mask the degree of enhancement on computed tomography (CT) scans. The choroid plexus is frequently enlarged with a positive correlation to the extent of the leptomeningeal angioma (Figure 2). The underlying white matter typically shows accentuated T2 shortening.7
Unilateral cerebral hemisphere involvement eventually becomes atrophic, with dystrophic calcifications in most cases (Figure 3). Cranial asymmetry often results from the cerebral hemiatrophy. There is thickening of the ipsilateral calvarium and enlargement of the paranasal sinuses and mastoid air cells owing to the lack of brain growth on the affect side.
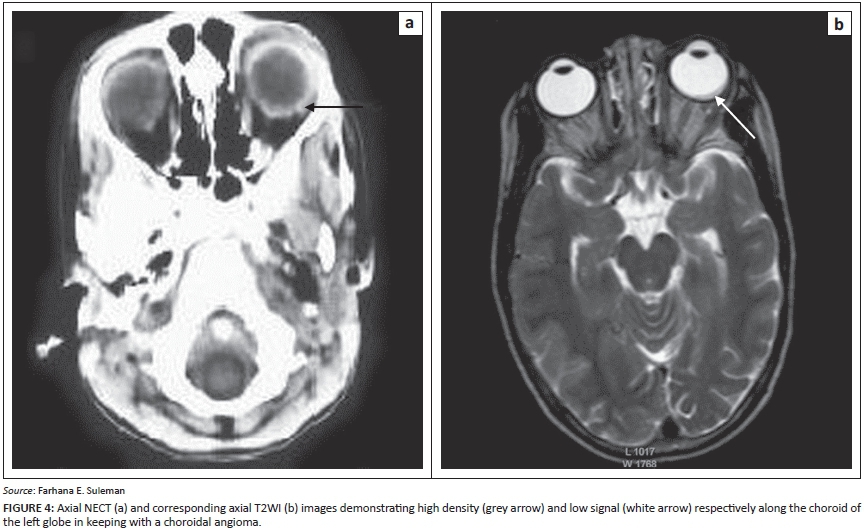
About 30% of patients with SWS have angiomas of the choroid and sclera (Figure 4). The presence of choroidal angioma correlates with the presence of bilateral disease and the extent of facial involvement; it does not correlate with the size of the intracranial pial angioma.
Conclusion
Bilateral intracranial calcification in SWS has been described only occasionally, and is not commonly referred to in the widely used radiology, neurology or paediatric texts. The clinical correlation of intractable seizures with the presence of bilateral intracranial disease has management and prognostic implications, thus making the presence of bilateral disease an important factor to all those involved in the management of the child with SWS.
Competing interests
The authors declare that they have no financial or personal relationships which may have inappropriately influenced them in writing this article.
Authors' contributions
N.A. (University of Pretoria) wrote the original manuscript. J.S. (University of Pretoria) identified the cases and contributed to the literature review. F.E.S. (University of Pretoria) prepared the images and was responsible for final editing of the report.
References
1.Takeoka M, Riviello Jr JJ. Sturge-Weber syndrome. Medscape. 05 January 2010 [cited 2011 Jan 11]. Available from: http://www.Emedicine.medscape.com/article/1177523-overview [ Links ]
2. Bolthauser E, Wilson J, Hoare RD. Sturge-Weber syndrome with bilateral intracranial calcification. J Neurol Neurosurg Psychiatry. 1976;39:429-435. http://dx.doi.org/10.1136/jnnp.39.5.429 [ Links ]
3. Evans AL, Widjaja E, Connolly DJA, Griffiths PD. Cerebral perfusion abnormalities in children with Sturge-Weber syndrome shown by dynamic contrast bolus magnetic resonance perfusion imaging. Pediatrics. 2006;117:2119-2125. http://dx.doi.org/10.1542/peds.2005-1815 [ Links ]
4. Hoffman HJ, Hendrik EB, Dennis M, Armstrong D. Hemispherectomy for Sturge-Weber syndrome. Childs Brain.0 1979;5:233-248. http://dx.doi.org/10.1159/000119821 [ Links ]
5. Rappaport ZH. Corpus callosum section in the treatment of intractable seizures in the Sturge-Weber syndrome. Childs Nerv Syst. 1988;4:231-232. http://dx.doi.org/10.1007/BF00270919 [ Links ]
6. Griffiths PD, Boodram MB, Blaser S, Armstrong D, Gilday DL, Harwood-Nash D. 99mTechnetium HMPAO imaging in children with the Sturge-Weber syndrome: A study of nine cases with CT and MRI correlation. Neuroradiology. 1997;39: 219-224. http://dx.doi.org/10.1007/s002340050398 [ Links ]
7. Barkovich JA. Pediatric neuroimaging. 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2005; p. 476-481. [ Links ]
 Correspondence:
Correspondence:
Farhana Suleman
Private Bag X169, Pretoria 0001
South Africa
Email: fesuleman@gmail.com
Received: 28 Nov. 2014
Accepted: 11 Apr. 2015
Published: 14 Aug. 2015


{kind=link}
{kind=link}
{kind=link}