










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSA Journal of Radiology
versão On-line ISSN 2078-6778
versão impressa ISSN 1027-202X
S. Afr. J. radiol. (Online) vol.18 no.2 Johannesburg 2014
http://dx.doi.org/10.4102/sajr.v18i2.703
CASE REPORTS
Secondary chondrosarcoma: Malignant transformation of pre-existing hereditary and non-hereditary cartilaginous lesions
Susanna C.S. VlokI; Georg W.W. WagenerI; Dan ZaharieII
IDivision of Radiodiagnosis, Stellenbosch University and Tygerberg Hospital, South Africa
IIDepartment of Anatomical Pathology, Stellenbosch University and Tygerberg Hospital, South Africa
ABSTRACT
Secondary chondrosarcoma is a malignant hyaline cartilage tumour originating from a cartilaginous precursor, either osteochondroma or enchondroma. We contrast two different cases of biopsy-proven secondary chondrosarcomas resulting from benign, pre-existing cartilaginous lesions - our aim is to contrast and compare these two benign conditions consisting of multiple cartilaginous lesions - one hereditary and the other non-hereditary - and emphasise their potential for malignant transformation.
Introduction
Chondrosarcoma is a malignant cartilaginous tumour of either primary or secondary origin.1 Primary chondrosarcoma originates in previously normal bone in the absence of a precursive cartilaginous lesion1,2,3 and is more common than secondary chondrosarcoma2 which results as a complication of a pre-existing cartilaginous lesion.3
Case reports
Case 1
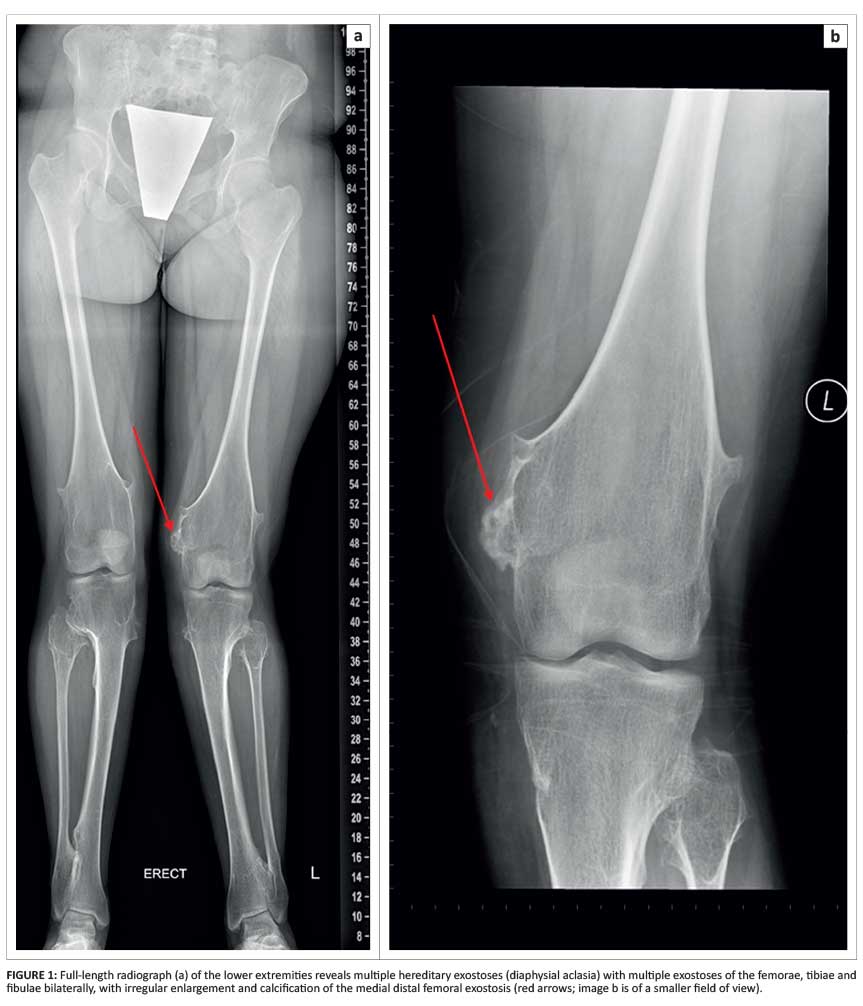
An 18-year-old girl presented with bilateral bony knee deformities and associated pain. A full-length radiograph of the lower extremities demonstrated multiple sessile and pedunculated exostoses bilaterally. Peripheral permeation and heterogenous appearance of the medial distal femoral exostosis was noted (Figure 1).
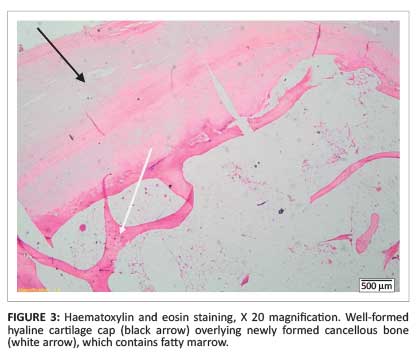
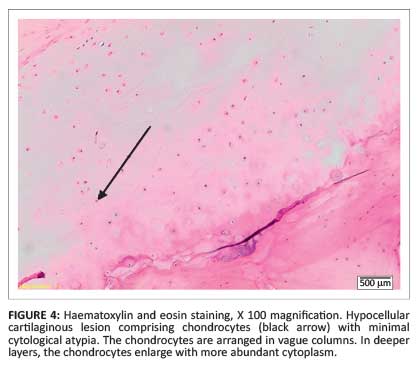
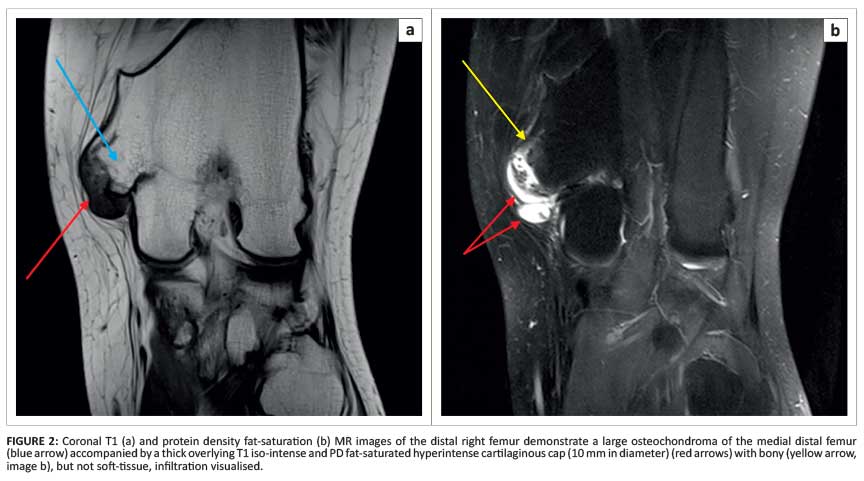
Magnetic resonance imaging (MRI) showed a large osteochondroma of the medial distal femur covered by an irregular lobular cartilage cap with a maximum diameter of 10 mm as well as underlying T2 hyperintense bone changes (Figure 2), suggestive of malignant transformation. Histological analysis confirmed a low-grade chondrosarcoma (Figure 3 and Figure 4).
Case 2
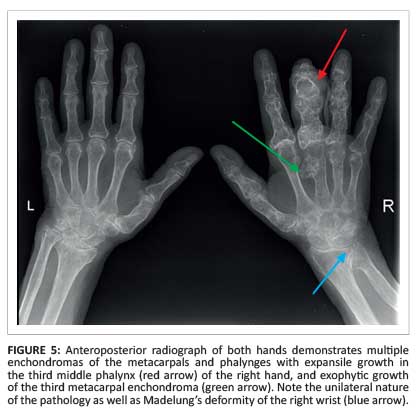
A 60-year-old woman with known Ollier disease presented with increased swelling and pain of the right middle finger. Conventional radiography of the hands showed multiple enchondromas unilaterally in the right hand (Figure 5).
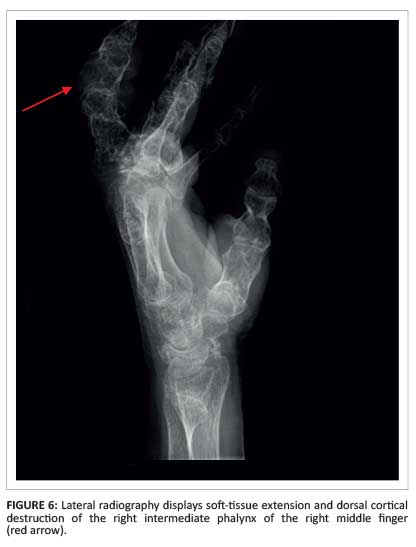
Cortical destruction of the intermediate phalanx of the right middle finger, with an associated soft-tissue mass containing chondroid calcifications (Figure 6), suggested malignant transformation.
Histology obtained after surgical amputation of the third finger revealed a grade II chondrosarcoma (Figure 7 and Figure 8).
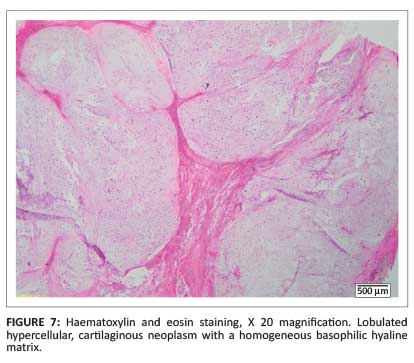
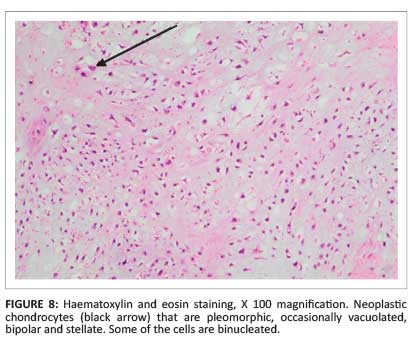
Discussion
Multiple hereditary osteochondromas/exostoses (diaphyseal aclasis)
Osteochondroma, the most common benign cartilaginous tumour, is a cartilage-capped bony exostosis arising from the cortical surface of bone. The cortex is uninterrupted from the underlying bone, and the marrow cavity is continuous with the native bone.3,4 A diagnosis of multiple hereditary osteochondroma can be made when at least two osteochondromas are detected radiologically in the juxta-epiphyseal region of long bones. A positive family history is obtained in only 60% of cases.4 Multiple hereditary osteochondromas is an uncommon, autosomal dominant condition with a prevalence of 1:50 000 - 1:100 000 people4 and a slight male predominance.3
Osteochondromas develop shortly after birth in the metaphyses of long bones, in close proximity to the epiphyseal plates, and are usually discovered around the median age of three years - almost all by the age of 12 years - when patients present with bony deformities, nerve compression and pain.4,5 Malignant transformation of these exostoses is much higher than in sporadic cases (where it is less than 1%), and has been found to be as high as 25% (lower rates of 3% - 5% have also been published).6 Lesions typically form and enlarge during growth until the skeleton is fully matured. Osteochondromas are more commonly broad-based and sessile and less often pedunculated.2 Lesions are typically multiple and bilateral, and most commonly affect the knee, humerus, hip and, to a lesser extent, the wrist, ankle and ribs.3
Radiographs of the sessile form show a well-defined bulging cortex associated with normal underlying bone. Lesions tend to be symmetric and associated with several bone deformities including bilateral coxa valga. In the pedunculated form, a stalk arises from normal underlying bony cortex and medulla, projecting away from the joint line, growing along the direction of the tendons. MRI of exostoses demonstrates a cortex which is continuous with that of the underlying bone, covered by a T2 bright cartilage cap, with no soft-tissue mass or underlying native bony abnormalities. Ultrasound can be used to evaluate and measure the cartilaginous cap diameter if the lesion is superficial.
Radiographic features suggesting malignant transformation to chondrosarcoma include osseous destruction, scattered calcifications, irregularity and lobulation of the cartilage cap (as demonstrated by the patient described in case 1) and a cartilaginous cap diameter exceeding 10 mm.1,2,4 Careful follow-up and monitoring of the size of osteochondromas (in particular, those involving the pelvis or scapula which are known to have an increased tendency to malignant transformation) may aid in early identification of secondary chondrosarcoma.5
Multiple enchondromatosis (Ollier disease)
An enchondroma is a benign cartilaginous lesion originating from the medullary cavity. Enchondromas are the result of the continued growth of residual benign cartilaginous rests owing to failure of cartilage ossification.7
Multiple enchondromatosis (Ollier disease) is a rare developmental abnormality with an incidence of 1:100 000 people.8 The disease usually appears in early childhood, is neither hereditary nor familial, and is considered to be a dysplasia. It is characterised by the presence of enchondromas in the metaphyses and diaphyses of multiple bones, with a tendency to be unilateral and localised to one extremity (as with our patient in case 2).7
Patients usually present with unilateral, multiple, painless, palpable bony masses on the fingers or toes, associated with shortening of extremities on the affected side and mildly delayed bone age. Enchondromas that enlarge after puberty are suggestive of malignant transformation, especially if associated with pain and pathological fractures.8
Radiographic and computed tomographic (CT) examinations reveal multiple lucent lesions with narrow zones of transition, in the absence of periosteal reactions, cortical breakthrough or soft-tissue involvement. Chondroid calcifications in the form of rings and arcs may be present.7,8 Lesions may sometimes be large and even grotesque, especially in the fingers.2 On MRI, enchondromas appear as intra-medullary, well-circumscribed lesions with intermediate to low T1 signal, high T2 signal and no surrounding oedema.2,7,8,9
The most severe complication of Ollier disease is malignant transformation to chondrosarcoma,2 which is seen in approximately 25% - 30% of cases by the age of 40 years.10 This happens in less than 1% of sporadic cases.9 Radiological features suggestive of malignant transformation include cortical breach, scalloping involving more than two-thirds of the surrounding cortex, an associated soft-tissue mass, and lesions localised to the spine, pelvis, sacrum, ribs and sternum. Distinguishing enchondromas from low-grade chondorsarcomas may prove a diagnostic dilemma as both these entities may have similar radiological and histological appearances.7,8
Treatment of Ollier disease consists of intervention to treat deformities and surveillance for malignant transformation. Malignancy typically requires surgical intervention.7
Teaching point
Exostoses of multiple hereditary osteochondromas and enchondromas owing to Ollier disease undergo malignant transformation to chondrosarcomas in about 25% of cases whereas sporadic exostoses and enchondromas undergo malignant transformation in less than 1% of cases.
Conclusion
Rare, pre-existing cartilaginous lesions such as multiple hereditary osteochondromas and Ollier disease may undergo malignant transformation to chondrosarcoma. Malignant transformation occurs in up to 25% of cases, especially if the lesions are located in the pelvis or scapula.
Ollier disease is a non-hereditary condition consisting of multiple enchondromas. Enlargement of enchondromas post puberty, especially when associated with pain, raises the suspicion of malignant transformation, which happens in 25% - 30% of cases. Lesions localised to the spine, pelvis, sacrum, ribs and sternum carry a high probability for malignant transformation.
Acknowledgements
Competing interests
The authors declare that they have no financial or personal relationship(s) that may have inappropriately influenced them in writing this article.
Authors' contributions
S.C.S.V. (Stellenbosc
References
1. Manaster BJ. Chondrosarcoma. STATdx Premier.Amirsys; 2005-2014. [ Links ] [cited 2014 May 20-25]. Available from: http://my.statdx.com
2. Manaster BJ, May DA, Disler DG. The requisites, musculoskeletal imaging. 3rd edn. Philadelphia: Mosby; 2007; p. 445, 477-488. [ Links ]
3. Dähnert W. Radiology review manual. 6th edn. Philadelphia: Lippincott Williams & Wilkins; 2007; p. 58-60, 133-135. [ Links ]
4. Bovée JVMG. Multiple osteochondromas. Orphanet J Rare Dis. 2008;3:3. http://dx.doi.org/10.1186/1750-1172-3-3 [ Links ]
5. Wuyts W, Schmale GA, Chansky HA, Raskind WH.Hereditary multiple osteochondromas. GeneReviews [homepage on the Internet]. [ Links ] [Updated 2000 Aug 3; cited 2013 Nov 21]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1235
6. Lee JK, Yao L, Wirth CR. MR imaging of solitary osteochondromas: Report of eight cases. AJR. 1987;149(3):557-560. http://dx.doi.org/10.2214/ajr.149.3.557 [ Links ]
7. Murphey MD, Flemming DJ, Boyea SR, Bojescul JA, Sweet DE, Temple HT. Enchondroma vs chondrosarcoma in the appendicular skeleton: Differentiating features. Radiographics. 1998;18(5):1213-1237. http://dx.doi.org/10.1148/radiographics.18.5.9747616 [ Links ]
8. Thomas AJ. Multiple enchondromatoses. STATdx Premier. Amirsys; 2005-2014. [ Links ] [cited 2014 May 20-25]. Available from: http://my.statdx.com
9. Bullough PG. Orthopedic pathology. 4th ed. Philadelphia: Mosby; 2004; p. 405-408, 416. [ Links ]
10. Gabos PG, Bowen JR. Epiphyseal-metaphyseal enchondromatosis. A new clinical entity. J Bone Joint Surg Am. 1998;80(6):782-792. [ Links ]
 Correspondence:
Correspondence:
Susanna Vlok
11 Bergzicht, Wylant Street
Welgemoed, Bellville 7530
South Africa
Email:sucarivlok@gmail.com
Received: 28 Jul. 2014
Accepted: 20 Nov. 2014
Published: 15 Dec. 2014


{kind=link}
{kind=link}