










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSA Journal of Radiology
On-line version ISSN 2078-6778
Print version ISSN 1027-202X
S. Afr. J. radiol. (Online) vol.18 n.2 Johannesburg 2014
http://dx.doi.org/10.4102/sajr.v18i2.667
CASE REPORTS
Andrew J. van den Heever
Tuft and Partners Incorporated Radiologists, Cape Town, South Africa
ABSTRACT
Macrodystrophia lipomatosa (MDL) is a rare congenital, but non-hereditary, form of localised gigantism of the fingers or toes. The hallmark of the condition is hypertrophy of all mesenchymal elements in the involved region, with a predominance of fibro-adipose tissue. A clinically subtle case of MDL is presented.
Introduction
A clinically subtle case of macrodystrophia lipomatosa (MDL) of the left hand is presented, with emphasis on the characteristic magnetic resonance imaging (MRI) features of the condition that assisted in the radiological diagnosis.
Case presentation
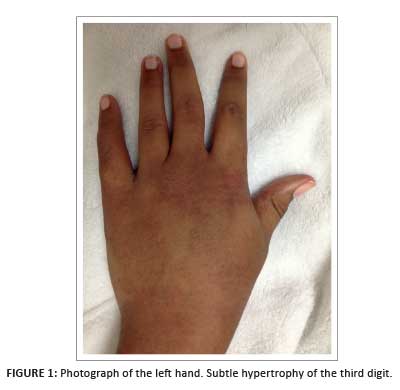
A 16-year-old girl presented with a swollen, erythematous and warm left hand of long-standing duration (Figure 1). The initial clinical suspicion included cellulitis or a retained foreign body. There was no trophic skin change, and the neuromuscular examination of the limb was normal.
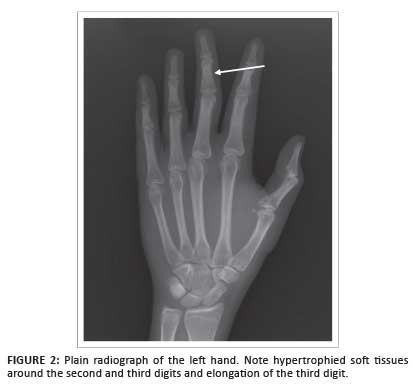
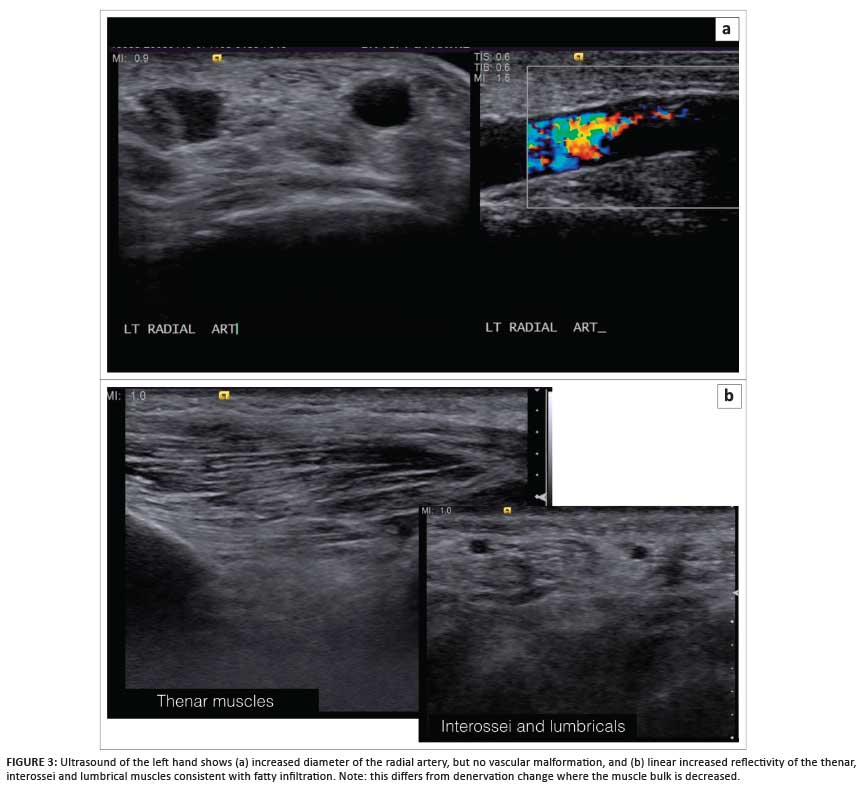
Following a plain radiograph of the left hand (Figure 2), an ultrasound scan was requested; a diagnosis of MDL was suggested, based on the findings of excessive fatty and vascular elements within a region of relative hypertrophy matching that of the median nerve sclerotome (Figure 3).
Magnetic resonance imaging (MRI) confirmed the diagnosis and excluded vascular or lymphatic malformation. The patient was scanned with both hands and wrists together (palms facing) to demonstrate the asymmetrical fatty and vascular hypertrophy (Figures 4-7.)
Discussion
MDL is a rare congenital, but non-hereditary, form of localised gigantism of the fingers or toes with progressive proliferation of all mesenchymal elements in the distribution of a sclerotome and a disproportionate increase in fibro-adipose tissue.1,2,3 It can involve both the upper extremities (usually in the distribution of the median nerve with the index and middle fingers most commonly involved) and the lower extremities (usually in the distribution of the medial plantar nerve).4 MDL is mostly unilateral, with lower limb involvement more common.
Accelerated soft-tissue and osseous growth in the involved region is self-limiting and usually terminates at puberty.5 MDL may be associated with syndactyly, polydactyly, brachydactyly or clinodactyly.4
The hallmark of MDL is hypertrophy of all mesenchymal elements in the involved region, with a predominance of fatty tissue. Fibro-adipose tissue is produced in association with localised hyperplastic vascular and lymphatic elements.6
The pathologic findings are infiltration and hypertrophy of adipose tissue in subcutaneous tissue, nerve sheaths and periosteum.5 Proposed aetiologies include disturbed foetal circulation, trophic influence of a tumified nerve, in utero disturbance of growth-limiting factor, or an error in segmentation.7
The differential diagnoses include neurofibromatosis type 1, vascular malformation, fibrolipomatous hamartoma of the median nerve, Klippel-Trenaunay-Weber syndrome and hemihypertrophy (Proteus and Beckwith-Wiedemann syndromes).8
Treatment goals are to (1) maintain nerve and joint function by minimising local tissue trauma; and (2) improve cosmesis. Surgical management includes inter alia debulking, epiphyseodesis and osteotomy.
Plain radiographs usually demonstrate the osseous overgrowth in the involved digits. However, in cases such as the present, where the hypertrophy is not marked, it may be overlooked. This case also demonstrates how the fatty overgrowth typical in MDL may be confusing on ultrasound examination. The diffuse fatty hypertrophy in the subcutaneous tissues and within the intrinsic musculature all appears diffusely echogenic, which results in a loss of soft-tissue detail and a decrease in the extent of grey-scale difference (reflectivity) between soft-tissue structures.
The key to making the diagnosis on ultrasound is recognising the abnormal tissue echogenicity consistent with increased fat. MRI is the investigation of choice owing to its superior soft-tissue contrast and its ability to characterise soft tissue, particularly fat. The contrast between fat and muscle allows effective visualisation of fatty infiltration of muscle; this can be confirmed with fat suppression techniques. Increased vascularity is also clearly visualised as vascular flow-voids or by performing MR angiography.
Conclusion
MDL is a rare congenital form of localised gigantism. In cases such as the present, where the hypertrophy of involved tissues is not marked, the condition may be missed or incorrectly diagnosed. In such cases, MRI is the investigation of choice owing to its ability to differentiate and characterise soft-tissue structures. Ultrasound is useful in excluding conditions included in the clinical differential diagnosis, but may be misleading owing to the uniform echogenicity of structures infiltrated by fat.
Acknowledgements
Competing interests
The author declares that they he has no financial or personal relationships that he might have inappropriately influenced him in writing this article.
References
1. Singla V, Virmani V, Tuli P, Singh P, Khandelwal N. Case Report: Macrodystrophia lipomatosa - Illustration of two cases. Indian J Radiol Imaging. 2008; 18(4): 298- 301. http://dx.doi.org/10.4103/0971-3026.43844 [ Links ]
2. Suleman FE, Kisansa M. Imaging of a rare disorder: macrodystrophia lipomatosa. S Afr J Rad. 2010; 14(2): 39-42. [ Links ]
3. Aga P, Parashari UC, Parihar A, et al. Macrodystrophia lipomatosa - MR imaging of a rare congenital anomaly: review of 3 cases. S Afr J Rad. 2010; 14(4): 105-107. [ Links ]
4. Khan RA, Wahab S, Ahmad I, Chana RS. Macrodystrophia lipomatosa: four case reports. Ital J Pediatr. 2010; 36:69. http://dx.doi.org/10.1186/1824-7288-36-69 [ Links ]
5. Mannan R, et al. Cyto-Radiological Diagnosis of Macrodystrophia Lipomatosa: A Report of Rare Entity With Review of Literature. Int J Med Health Sci. 2013; 2(4): 469-471. [ Links ]
6. Murphey MD, Carroll JF, Flemming DJ et al. From the archives of the AFIP: benign musculoskeletal lipomatous lesions. Radiographics 2004; 24(5): 1433-1466. [ Links ]
7. Blacksin M, Barnes FJ, Lyons MM. MR diagnosis of macrodystrophia lipomatosa. AJR Am J Roentgenol 1992; 158(6): 1295-1297. [ Links ]
8. Rohilla S, Jain N, Sharma R, Dhaulakhandi DB. Macrodystrophia lipomatosa involving multiple nerves. J Orthop Traumatol. 2012; 13(1): 41-45. http://dx.doi.org/10.1007/s10195-011-0159-6 [ Links ]
 Correspondence:
Correspondence:
Andrew van den Heever
PO Box 461, Plumstead 7801
South Africa
Email:andrew.vdh@tuft.co.za
Received: 09 Jun. 2014
Accepted: 10 Nov. 2014
Published: [to be released]


{kind=link}