










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSA Journal of Radiology
versão On-line ISSN 2078-6778
versão impressa ISSN 1027-202X
S. Afr. J. radiol. (Online) vol.18 no.1 Johannesburg Out. 2014
http://dx.doi.org/10.4102/sajr.v18i1.602
PICTORIAL ESSAY
Intramedullary spinal masses: A pictorial essay
Abraham Fourie Bezuidenhout; Jared M. Lipsitz; Anne-Marie Du Plessis
Department of Diagnostic Radiology, Stellenbosch University, South Africa
ABSTRACT
This pictorial essay provides a classification system for the most common intramedullary spinal masses and describes distinguishing imaging features that will aid the radiologist in providing a relevant differential diagnosis to guide further management.
Introduction
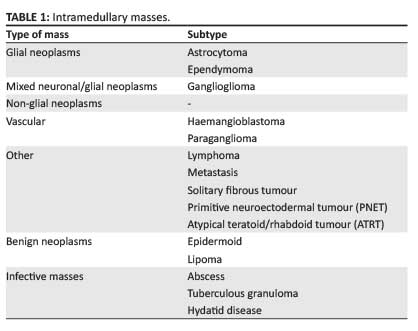
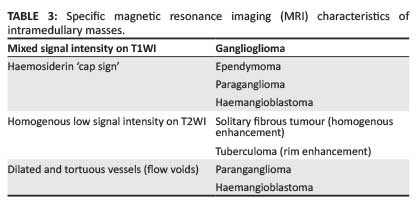
The clinical symptoms of intramedullary spinal masses, such as back pain, radiculopathy and myelopathy, are nonspecific; therefore responsibility falls on the radiologist to provide a relevant differential diagnosis. The imaging gold standard for the evaluation of intramedullary masses is magnetic resonance imaging (MRI). On MRI, almost all intramedullary neoplasms expand the spinal cord, with variable post-contrast enhancement; absence of a combination of these features should suggest a non-neoplastic aetiology.1 Further associated imaging findings include spinal canal remodeling, syrinx formation, and tumoural and non-tumoural cysts.2 The most common differential diagnoses to be considered for intramedullary masses are malignant neoplasms (glial and non-glial tumours), benign neoplasms and, considering the large HIV-positive population, infective aetiologies. Spinal cord ependymomas are the most common intramedullary neoplasms in adults, and astrocytomas in children; combined, they constitute 70% of intramedullary neoplasms.1The aforementioned neoplasms, together with gangliogliomas, comprise the most common intramedullary spinal glial tumours. Non-glial neoplasms include vascular tumours such as haemangioblastomas and paragangliomas, and less common neoplasms such as lymphoma, primitive neuroectodermal tumours (PNETs), solitary fibrous tumours and metastases. Benign neoplasms include epidermoids and lipomas (Table 1).3,4
Glial neoplasms
Ependymoma
Ependymomas are the most common intramedullary spinal neoplasms in adults.5 Most of these are low-grade neoplasms (i.e. World Health Organization [WHO] classification grade I or grade II). Histologically, the cellular subtype is classic and the most common (Figure 1).1 As these neoplasms arise from the ependymal cells lining the central canal, they are located centrally in the spinal cord, grow concentrically outward, are well circumscribed and demonstrate intense homogenous post-contrast enhancement. Cystic degeneration is seen in 50% of cases and haemorrhage is common at the superior and inferior margins; this causes the so-called haemosiderin 'cap sign' identified as hypointensity on T2-weighted images (T2WI) seen in about 20%-33% of cases.1,5

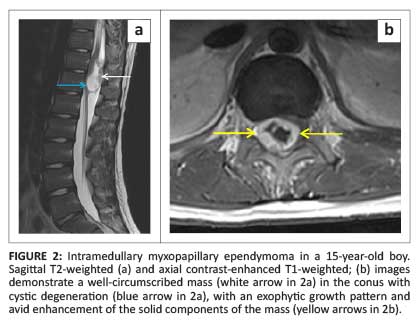
Six histological subtypes are recognised; of note is the myxopapillary subtype which has a predilection for the conus medullaris and filum terminale, and represents the most common conus neoplasm. These neoplasms occur predominantly in young men and may occasionally demonstrate high signal intensity on both T1WI and T2WI, a finding that reflects mucin content or haemorrhage.1 However, the imaging findings are generally non-specific and the conus medullaris location should be suggestive of the diagnosis (Figure 2).
Astrocytoma
Astrocytomas arise from astrocytic glial cells and are the most common intramedullary tumours in children.2 These are usually low-grade neoplasms, with less than 2% being glioblastomas (Figure 3).5
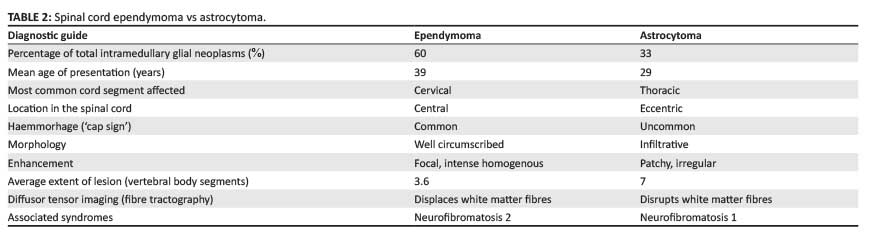
Unlike their intracranial counterparts, spinal astrocytomas often demonstrate patchy post-contrast enhancement. They are usually eccentrically located within the cord and ill-defined owing to their infiltrative growth pattern. As cerebrospinal fluid (CSF) dissemination may occur, imaging of the entire neuraxis is recommended.2 Diffusor tensor imaging (fiber tractography) has been used to differentiate ependymomas, which displace rather than infiltrate white-matter tracts, from astrocytomas that disrupt the descending fibres (Figure 4).5,6 It is very difficult to distinguish ependymomas from astrocytomas on imaging, but Table 2 provides some detail that might help to simplify the process.
Mixed neuronal/ glial neoplasm
Ganglioglioma
Gangliogliomas are low-grade neoplasms that occur in people at an average age of 19 years, and represent a mixture of neuronal elements (ganglion cells) and glial elements (primarily neoplastic astrocytes).1 A study by Patel et al. found that 84% of gangliogliomas had mixed signal intensity on T1WI (which is a unique finding for spinal neoplasms) and speculated that this was because of the dual cellular population.7 These neoplasms may involve long segments of the cord (commonly extending more than eight vertebral body segments), contain calcifications and often have very little surrounding oedema.8
Non-glial neoplasm: Vascular neoplasms
Haemangioblastoma
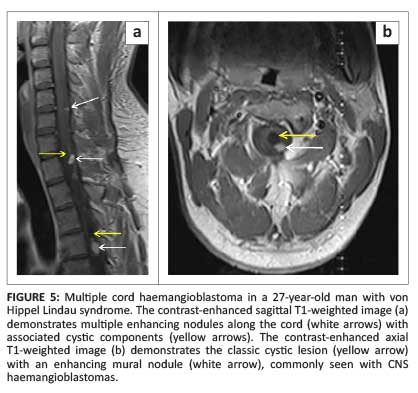
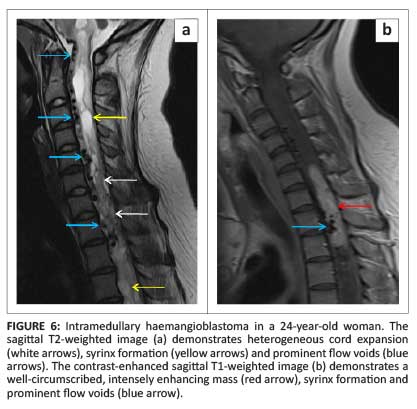
Most cord haemangioblastomas (80%) are solitary with multiple lesions indicative of von Hippel-Lindau syndrome (Figure 5a).1 These neoplasms are capillary-rich nodular masses abutting the leptomeninges, with prominent dilated and tortuous vessels on the cord surface (Figure 6). Their appearance may reflect the classic cystic lesion with an enhancing mural nodule, commonly seen in their intracranial counterparts (Figure 5b). Syrinx and spinal cord oedema are characteristic findings even in small tumours.8 Imaging of the entire neuraxis is recommended for patients with a positive family history of von Hippel-Lindau syndrome.
Paraganglioma
Paragangliomas are neuroendocrine neoplasms arising from paraganglia. Paraganglia are accessory organs of the peripheral nervous system.1 They are encapsulated, highly vascular neoplasms with a predilection for the conus medullaris, cauda equina and filum terminale.8 Haemorrhage produces the 'cap sign', and intratumoural vessels with flow voids are common features of this tumour. Occasionally the characteristic 'salt-and-pepper' appearance, similar to neck and skull base paragangliomas, may be seen.
Rare non-glial neoplasm
Lymphoma
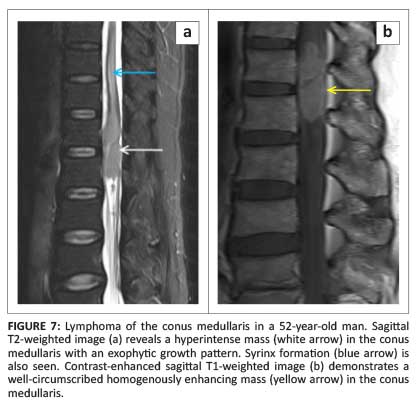
Primary intramedullary lymphomas are rare, comprising 3.3% of central nervous system (CNS) lymphomas.9 There are only a handful of reports on the MRI findings of intramedullary lymphomas that describe high signal intensity on T2WI in contrast to the low signal intensity found in intracranial lesions and non-specific post-contrast enhancement (Figure 7).1
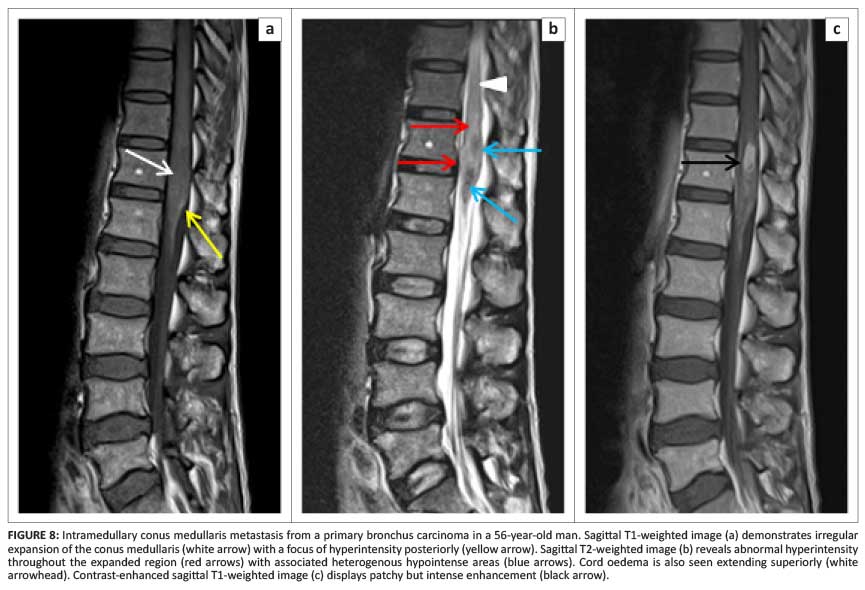
Metastasis
Intramedullary metastases are rare and most commonly secondary to bronchus and breast carcinoma (Figure 8).1 Cerebellar medulloblastoma has also been known to cause intramedullary metastasis.1 Spread may either be haematogenous or from the leptomeninges. Intramedullary metastases show avid post-contrast enhancement and profuse oedema which is often disproportionately extensive compared with the size of the lesion.8 Associated cysts are rare, compared with primary neoplasms. As cord expansion may be mild, differentiating intramedullary metastasis from demyelinating conditions can be difficult.
Solitary fibrous tumour
Solitary fibrous tumours (SFTs) are uncommon spindle-cell neoplasms that typically arise in the pleural cavity. The first CNS SFT was described in 1996 and has received great attention since then.10 These tumours typically occur in adults and are usually well-circumscribed and show homogenous enhancement post contrast. Until this point, we have not dwelled on signal characteristics of the described conditions, as most spinal cord neoplasms are hypo- to isointense on T1WI and hyperintense on T2WI, offering no assistance in differentiating individual intramedullary neoplasms. SFTs, however, demonstrate hypointense signal on T2WI, clearly defining this from other intramedullary neoplasms (Figure 9). This unique imaging appearance is caused by the hypocellularity of, and abundant collagen stroma within this tumour.11

Primitive neuroectodermal tumour
Intramedullary primitive neuroectodermal tumour (PNET) is a rare, aggressive embryonal tumour and is more commonly seen secondary to metastatic spread through the CSF from an intracranial primary tumour. Intramedullary PNET is slightly more common in young adults than in children.2 MRI findings are non-specific including T1 hypointensity, T2 hyperintensity and avid post-contrast enhancement.
Atypical teratoid/rhabdoid tumour
Atypical teratoid/rhabdoid tumour (ATRT) is a rare intramedullary tumour that mainly occurs in children less than 2 years old, and shares MRI characteristics with PNETs.2
Benign neoplasms
Epidermoid
Intramedullary epidermoids are ectodermal inclusion neoplasms that result from anomalous implantation during neural tube closure.12 Progressive desquamation of keratin from the epithelial lining into the interior of the neoplasm produces a cystic lesion that follows CSF intensity on T1- and T2-weighted sequences but demonstrates characteristic 'dirty' or heterogenous hyperintense fluid-attenuated inversion recovery (FLAIR) signal and restricted diffusion on diffusion-weighted imaging, which aids in differentiating this neoplasm from others.13
Lipoma
Intramedullary lipomas consist of normal fat. The pathophysiology of these lesions is poorly understood. They usually occur along the dorsal midline of the spinal cord but rarely infiltrate the cord.14 They follow fat signal on all sequences and suppress on fat-supression sequences.
Infective masses
Abscess
Intramedullary suppuration of the spinal cord is rare. Diagnosis of an intramedullary abscess should be considered in cases of acute neurological deterioration referable to spinal cord fall-out. CSF analysis plays an important role in differentiating between neoplastic and infective masses on initial imaging.8 Chronological MRI changes in intramedullary abscesses are similar to those in the brain. In the early stages of infectious myelitis, MRI demonstrates hyperintensity on T2WI with poorly defined enhancement on post-contrast T1WI; ultimately the mass becomes less diffusely hyperintense on T2WI, with more clearly defined marginal post-contrast enhancement.15
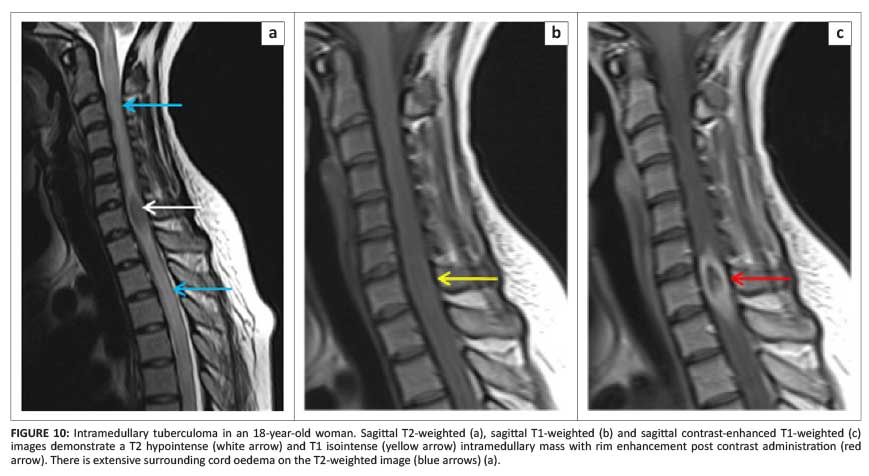
Tuberculous granuloma
Tuberculous granuloma (tuberculoma) needs special mention owing to its characteristic caseating solid centre demonstrating hypo- to isointense signal on both T1- and T2WI with post-contrast rim enhancement (Figure 10).16
Hydatid disease
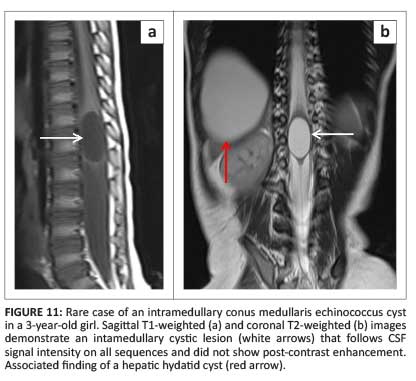
Although intramedullary hydatid disease is extremely rare, owing to its endemicity, it should be considered in the differential of a cystic intramedullary mass. Spinal cord hydatid disease follows CSF signal intensity on all sequences but differs from its intracranial counterparts in that it does not demonstrate rim enhancement post contrast and rarely calcifies (Figure 11).17
Acknowledgements
Competing interests
The authors declare that they have no financial or personal relationship(s) that may have inappropriately influenced them in writing this article.
Authors' contributions
A.F.B. (Stellenbosch University), J.M.L. (Stellenbosch University) and A-M.d.P. (Stellenbosch University) contributed equally to the writing of this article.
References
1. Koeller KK, Rosenblum RS, Morrison AL. Neoplasms of the spinal cord and filum terminale: Radiologic-pathologic correlation. RadioGraphics. 2000;20(6):1721-1749. http://dx.doi.org/10.1148/radiographics.20.6.g00nv151721 [ Links ]
2. Smith AB, Soderlund KA, Rushing EJ, Smirniotopolous JG. Radiologic-pathologic correlation of pediatric and adolescent spinal neoplasms: Part 1, Intramedullary spinal neoplasms. Am J Roentgenol. 2012;198(1):34-43. http://dx.doi.org/10.2214/AJR.10.7311 [ Links ]
3. Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumors of the central nervous system. Acta Neuropathol. 2007;114(2):97-109. http://dx.doi.org/10.1007/s00401-007-0243-4 [ Links ]
4. Dähnert WF. Radiology Review Manual. Alphen aan den Rijn: Wolters Kluwer; 2007. [ Links ]
5. Yousem DM, Grossman RI. Neuroradiology, the requisites. Maryland Heights: Mosby; 2010. [ Links ]
6. Thurnher MM, Law M. Diffusion-weighted imaging, diffusion-tensor imaging, and fiber tractography of the spinal cord. Magn Reson Imaging Clin N Am. 2009;17(2):225-244. http://dx.doi.org/10.1016/j.mric.2009.02.004 [ Links ]
7. Patel U, Pinto RS, Miller DC, et al. MR of spinal cord ganglioglioma. Am J Neuroradiol. 1998;19:879-887. [ Links ]
8. Abul-kasim K, Thurnher MM, Mckeever P, Sundgren PC. Intradural spinal tumors: Current classification and MRI features. Neuroradiology. 2008;50(4):301-314. http://dx.doi.org/10.1007/s00234-007-0345-7 [ Links ]
9. Koeller KK, Smirniotopoulos JG, Jones RV. Primary central nervous system lymphoma: Radiologic-pathologic correlation. RadioGraphics. 1997;17:1497-1526 http://dx.doi.org/10.1148/radiographics.17.6.9397461 [ Links ]
10. Metellus P, Bouvier C, Guyotat J, et al. Solitary fibrous tumors of the central nervous system: Clinicopathological and therapeutic considerations of 18 cases. Neurosurgery. 2007;60(4):715-722. http://dx.doi.org/10.1227/01.NEU.0000255418.93678.AD [ Links ]
11. Kawamura M, Izawa K, Hosono N, Hirano H. Solitary fibrous tumor of the spinal cord: Case report and review of the literature. Neurosurgery. 2004;55(2):433. http://dx.doi.org/10.1227/01.NEU.0000130037.45768.84 [ Links ]
12. Gonzalvo A, Hall N, Mcmahon JH, Fabinyi GC. Intramedullary spinal epidermoid cyst of the upper thoracic region. J Clin Neurosci. 2009;16(1):142-144. http://dx.doi.org/10.1016/j.jocn.2008.04.017 [ Links ]
13. Chen S, Ikawa F, Kurisu K, et al. Quantitative MR evaluation of intracranial epidermoid tumors by fast fluid-attenuated inversion recovery imaging and echo-planar diffusion-weighted imaging. Am J Neuroradiol. 2001;22(6):1089-1096. [ Links ]
14. Wood BP, Harwood-Nash DC, Berger P, Goske N. Intradural spinal lipoma of the cervical cord. Am J Roentgenol. 1985;145(1):174-176. http://dx.doi.org/10.2214/ajr.145.1.174 [ Links ]
15. Iwasaki M, Yano S, Aoyama T, Hida K, Iwasaki Y. Acute onset intramedullary spinal cord abscess with spinal artery occlusion: A case report and review. Eur Spine J. 2011;20(Suppl 2):294-301. [ Links ]
16. Burrill J, Williams CJ, Bain G, Conder G, Hine AL, Misra RR. Tuberculosis: A radiologic review. Radiographics. 2007;27(5):1255-1273. http://dx.doi.org/10.1148/rg.275065176 [ Links ]
17. Polat P, Kantarci M, Alper F, Suma S, Koruyucu MB, Okur A. Hydatid disease from head to toe. Radiographics. 2003;23(2):475-494. http://dx.doi.org/10.1148/rg.232025704 [ Links ]
 Correspondence:
Correspondence:
Fourie Bezuidenhout
PO Box 19179,
Tygerberg 7505,
South Africa
Email: fouriebez@yahoo.com
Received: 05 Mar. 2014
Accepted: 02 Sept. 2014
Published: 17 Nov. 2014


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}