










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouthern African Journal of HIV Medicine
On-line version ISSN 2078-6751
Print version ISSN 1608-9693
South. Afr. j. HIV med. (Online) vol.15 n.2 Johannesburg Feb. 2014
CASE REPORT
Antiretroviral therapy-induced Leber's hereditary optic neuropathy
A MoodleyI; S BholaII; F OmarIII; J MogamberyIV
IMB ChB, FCP Neurol (SA), PhD, Department of Neurology, Grey's Hospital, Pietermaritzburg and University of KwaZulu-Natal, Durban, South Africa
IIMB ChB, Department of Neurology, Grey's Hospital, Pietermaritzburg and University of KwaZulu-Natal, Durban, South Africa
IIIMB ChB, MMed, FCPath (SA), Chemical Pathology Laboratory, Groote Schuur Hospital; National Health Laboratory Service and University of Cape Town, South Africa
IVMB ChB, FCP (SA), Department of Medicine, Grey's Hospital, Pietermaritzburg, South Africa
ABSTRACT
Optic neuropathy in HIV-infected patients results from the HIV infection itself, post-infectious auto-immune disease, opportunistic infections and drugs. Nucleoside reverse transcriptase inhibitors (NRTIs) such as zidovudine and stavudine have known mitochondrial toxicity and can cause mitochondrial myopathies, neuropathies, hyperlactataemia, and can induce mitochondrial genetic disorders. Individuals with the mutation for Leber's hereditary optic neuropathy (LHON), a mitochondrial disorder, are usually asymptomatic but develop visual loss when exposed to external triggers such as smoking. We report on two HIV-infected patients with LHON mutations (m.14484T>C and m.11778G>A) who developed profound visual loss with antiretroviral therapy. We postulate that the phenotypic expression of LHON in these genetically predisposed individuals was triggered by NRTI drugs lamivudine and tenofovir when used in combination, despite their relatively weak mitochondrial toxic effects.
Optic neuropathy in HIV disease is due to the virus itself, para-infectious disease, oppor-tunistic infections, compression, raised intra-cranial pressure or drug therapy.[1] Hence it is no surprise that visual loss from optic nerve disease is common in patients infected with HIV. Epidemiological studies are lacking, especially in South Africa (SA) where the prevalence of HIV is high.
The causes of HIV-associated optic neuropathies that are commonly encountered in clinical practice are listed in Table 1. The immune-mediated optic neuropathies are para-infectious and occur in the setting of seroconversion or as part of acute disseminated encephalomyelitis when CD4+ counts may be relatively normal; these often respond to intravenous steroid therapy. Infectious and lymphomatous optic neuropathies occur during advanced stages of immunocompromise when CD4+ counts are low; in such patients, identifying an infective cause is urgent to prevent blindness. Ethambutol-associated retrobulbar optic neuropathy is common, especially in disseminated tuberculosis where its prolonged use over 9 months is advocated.

The optic nerve is unique as it is a white matter tract that is particularly susceptible to physical, ischaemic, toxic and hypoxic insults due to: its relatively large diameter (1.2 million axons per nerve); its intra- and extra-cranial location; its myelinated and unmyelinated segments; and the high energy demand by the retinal ganglion cells.[2] The large accumulation of mitochondria within the optic nerve head is mostly responsible for its energy supply.[3] The function of these mitochondria is disturbed in the setting of Leber's hereditary optic neuropathy (LHON), where gene defects from base substitutions at 11778G>A, 14484T>C and 3460G>A cause optic nerve dysfunction in over 95% of patients by disrupting complex I-dependent adenosine triphosphate synthesis. The abnormal mitochondrial gene is maternally inherited with incomplete penetrance, as only 50% of males with the LHON mitochondrial mutation present with the disorder.[4] Expression of the disease occurs when triggered by environmental factors such as smoking, alcohol and acute illness. The typical presentation is sequential or bilateral, simultaneous central visual loss in the 2nd to 3rd decade of life in males carrying one of the LHON mitochondrial mis-sense mutations. Ten per cent of female carriers are symptomatic. There is initial disc swelling and circumpapillary telangiectasia, which within a few months are replaced by optic atrophy from axonal loss. Visual loss and centro-caecal scotomas progress over months to years. Idebenone, a co-enzyme Q10 derivative, has shown promise in delaying the progression of illness and in some cases substantially reversing the visual and field loss.[5]
We present two patients from our neuro-ophthalmology unit who presented with central visual loss after starting antiretroviral therapy containing tenofovir, lamivudine and efavirenz.
Patient 1
A 31-year-old HIV-infected man presented with a 4-month history of gradual and pro-gressive loss of bilateral vision. He denied having headaches, pain on eye movements or constitutional symptoms. He was previously unemployed. Two years before, he acquired pulmonary tuberculosis and was found to be HIV-infected; he was then prescribed tenofovir, lamivudine and efavirenz. He had a four pack-year smoking history, did not consume alcohol and had no family history of visual loss.
His visual acuity was 'counting fingers' on the right and 'hand movements' on the left. Central field defects were detected bi-laterally on confrontation, but were globally lost on automated Humphrey visual field (HVF) testing. Colour vision was severely impaired bilaterally (0/15 on Ishihara pseudo-isochromatic plates). Both pupils were sluggishly reactive and severe bilateral optic atrophy was present on funduscopy (Fig. 1). The visual evoked potential (VEP) P100 waveforms were obtainable bilaterally using goggles, but were markedly reduced in amplitude.
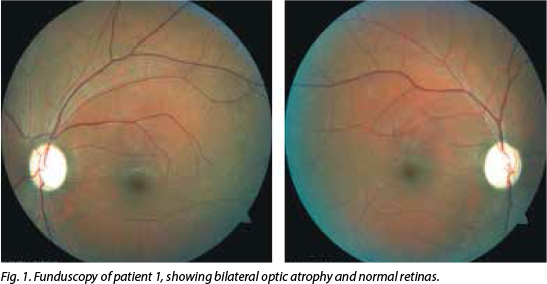
Magnetic resonance imaging (MRI) of the brain and orbits was normal. Chest radiograph and cerebrospinal fluid (CSF) exam-ination, including neurotropic virus (cyto-megalovirus, herpes simplex viruses 1 and 2, varicella zoster virus, Epstein-Barr virus and John Cunningham virus) testing were unremarkable and routine haematological investigations were normal. His vitamin B12, folate and iron levels were normal. CD4+ cell count was 746 cells/µl and viral load was undetectable. The antinuclear factor (ANF), antineutrophil cytoplasmic antibody (ANCA), rapid plasma reagin (RPR) and rheumatoid factor (RF) were negative, and serum angiotensin converting enzyme (SACE) was 19 U/l. Oligoclonal bands (OBs) were not detected in the CSF and aquaporin 4 antibodies were negative. He tested positive for the 14484T>C mitochondrial mutation associated with LHON.
Patient 2
A 33-year-old HIV-infected man who worked in construction presented with bilateral visual loss over the previous 3 years. He had been diagnosed with HIV infection after voluntary testing and prescribed tenofovir, lamivudine and efavirenz 3 months before the onset of his visual symptoms. He first noticed loss of central vision in the left eye, followed by similar symptoms in the right a month later. He denied any colour desaturation and pain on eye movements, but did notice intermittent oscillopsia at the onset of his symptoms. He had no other medical history of note and denied any smoking or alcohol use, exposure to any toxins or substance abuse. He had no family history of visual loss.
His visual acuity bilaterally was 'counting fingers'. Central scotomas were noted on con-frontation, but HVF showed bilateral, inferior altitudinal field defects. Colour vision was severely impaired (0/15 on Ishihara pseudo-isochromatic plates) and funduscopy revealed bilateral profound optic atrophy (Fig. 2). His pupils were reactive and a 1+ relative afferent pupillary defect was present on the right. The rest of his neurological examination was normal, and standard haematological, biochemical and CSF examination tests were normal. His CD4+ count was 615 cells/µl and viral load was 27 copies/ml. MRI of the brain and orbits was normal, as was his chest radiograph. The VEP P100 wave amplitudes were reduced bilaterally. His vitamin B12 level was mildly reduced at 117 pmol/l (normal 133 - 675), serum folate was normal and SACE was 27 U/l. ANF, ANCA, RF, RPR, human T lymphotropic virus 1, CSF OB and aquaporin 4 antibodies were negative. He tested positive for the 11778G>A mitochondrial mutation associated with LHON.
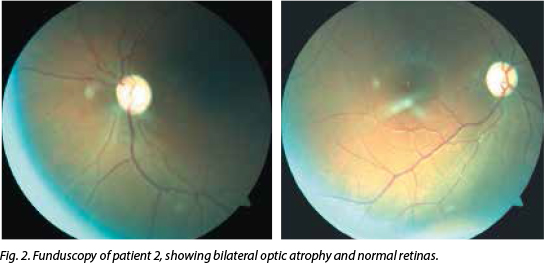
Discussion
Genetic testing of LHON by polymerase chain reaction and restriction enzyme analysis is offered by the Inherited Metabolic Disease Laboratory, National Health Laboratory Service, based at Groote Schuur Hospital in Cape Town. All six LHON mutations in mitochondrial DNA are screened for, viz. m.14484T>C, m.11778G>A, m.3460G>A, m.14459G>A, m.14482C>G, and m.14487T>C. Both patients had a common mitochondrial mutation associated with LHON. Patient 1 was a smoker without a significant pack-year history and patient 2 had mild vitamin B12 deficiency, neither of which seemed to have contributed significantly as triggers for LHON, or to the visual loss on a toxic or nutritional basis. Both patients, however, were HIV-positive, developed their symptoms some time after starting ART and were not on other medication that could have contributed to visual impairment.
LHON following the introduction of the strongly mitochondrial toxic nucleoside reverse transcriptase inhibitors (NRTIs) zidovudine, stavudine and zalcitabine (ddC) has previously been reported in patients with the mitochondrial mutations 11778G>A and 14484T>C.[6-8] No cases have been described with the weaker mitochondrial toxic NRTI drugs tenofovir and lamivudine until now. The catastrophic sequela of lactic acidosis that occurs with zidovudine and stavudine, however, is less likely to occur with the tenofovir-lamivudine combination. Tenofovir is a less toxic agent than zidovudine, didanosine, stavudine and zalcitabine, and does not usually cause lactic acidosis, myopathy and peripheral neuropathy. Its mechanism of action, like the other NRTIs, is mitochondrial DNA polymerase inhibition, and thus it is not devoid of toxic effects. Tenofovir-induced nephropathy occurs on the basis of mitochondrial toxicity within the kidneys' proximal tubular cells, which are rich in mitochondria.[9] Irregularly shaped mitochondria with fragmented cristae ensue. There are currently no data on the effects of tenofovir on the mitochondria of the optic neurons and retina. However, an analogous mechanism of toxicity at the optic nerve head, which also has abundant mitochondria, is conceivable with tenofovir. However, based on existing evidence, the role of lamivudine in mitochondrial toxicity is less clear. Hence, we postulate that in the genetically susceptible individual, tenofovir has the potential to trigger LHON.
Idebenone (co-enzyme Q10 derivative) is not available in SA. However, co-enzyme Q10 is available as an over-the-counter preparation at most pharmacies. Both patients were treated with this formulation and have not shown any deterioration or improvement after 6 months of treatment. ART was discontinued in both patients and when required, both patients will be considered for a combination of antiretrovirals with lower mitochondrial toxicities, such as protease and integrase inhibitors.
Conclusion
LHON can be triggered by NRTIs in HIV-infected patients who harbour the LHON mutations. The expression of LHON can occur regardless of the mitochondrial toxic potential of ART. The routine screening for LHON mutations on all asymptomatic male patients about to commence NRTIs is not cost effective. However, the progression of LHON is reasonably manageable with appropriate therapy; hence test-ing for LHON mutations in HIV-positive patients with optic neuropathy who are on ART should become standard practice.
References
1. Sudhakar P, Kedar S, Berger JR. The neuro-ophthalmology of HIV-AIDS. Neurobehav HIV Med 2012;4: 99-111. [http://dx.doi.org/10.2147/NBHIV.S24204] [ Links ]
2. Andrews RM, Griffiths PG, Johnson MA, Turnbill DM. Histochemical localisation of mitochondrial enzyme activity in human optic nerve and retina. Br J Ophthalmol, 1999;83(2):231-235. [http://dx.doi.org/10.1136/bjo.83.2.231] [ Links ]
3. Abu-Amero KK. Leber's hereditary optic neuropathy: The mitochondrial connection revisited. Middle East Afr J Ophthalmol 2011;18(1):17-23. [http://dx.doi.org/10.4103/0974-9233.75880] [ Links ]
4. Man PYW, Turnbull DM, Chinnery PF. Leber hereditary optic neuropathy. J Med Genet 2002;39(3):162-169. [http://dx.doi.org/10.1136/jmg.39.3.162] [ Links ]
5. Eng JG, Aggarwal D, Sadun AA. Idebenone treatment in patients with Leber hereditary optic neuropathy. Invest Ophthalmol Vis Sci 2009;50(Suppl):S1440. [ Links ]
6. Warner JE, Ries KM. Optic neuropathy in a patient with AIDS. J Neuroophthalmol 2001;21(2):92-94. [http://dx.doi.org/10.1097/00041327-200106000-00006] [ Links ]
7. Lüke C, Cornely OA, Fricke J, et al. Late onset of Leber's hereditary optic neuropathy in HIV infection. Br J Ophthalmol 1999;83(10):1204-1205. [http://dx.doi.org/10.1136/bjo.83.10.1194k] [ Links ]
8. Shaikh S, Ta C, Basham AA, Mansour S. Leber hereditary optic neuropathy associated with antiretroviral therapy for human immunodeficiency virus infection. Am J Ophthalmol 2001;131(1):143-145. [http://dx.doi.org/10.1016/S0002-9394(00)00716-9] [ Links ]
9. Abraham PRH. Depletion of the cellular antioxidant system contributes to tenofovir disoproxil fumarate-induced mitochondrial damage and increased oxido-nitrosative stress in the kidney. J Biomed Sci 2013;20(1):6. [http://dx.doi.org/10.1186/1423-0127-20-61] [ Links ]
 Correspondence:
Correspondence:
A Moodley
anand.moodley1@gmail.com

