










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Child Health
On-line version ISSN 1999-7671
Print version ISSN 1994-3032
S. Afr. j. child health vol.17 n.1 Pretoria 2023
http://dx.doi.org/10.7196/SAJCH.2023.v17i1.1779
RESEARCH
Clinical, biochemical, and histopathological diagnosis of Egyptian paediatric patients with suspected mitochondrial diseases: A hospital-based study
D M AbdouI; L A SelimII; R V CosterIII; J SmetIV; G A NakhlaV; D A MehaneyI
IMD; Clinical and Chemical Pathology Department, Faculty of Medicine, Cairo University, Egypt
IIMD; Pediatric Neurology Department, Faculty of Medicine, Cairo University, Egypt
IIIPhd; Department of Pediatrics, Division of Pediatric Neurology and Metabolism, Ghent University Hospital, Belgium
IVBSc; Department of Pediatrics, Division of Pediatric Neurology and Metabolism, Ghent University Hospital, Belgium
VMD; Pathology Department, Faculty of Medicine, Cairo University, Egypt
ABSTRACT
BACKGROUND: Mitochondrial respiratory chain (RC) disorders are a growing group of disorders with a large variety of clinical presentations ranging from well-defined clinical syndromes to nonspecific manifestations, such as failure to thrive, exercise intolerance and seizures
OBJECTIVE: To describe the clinical, biochemical, and histochemical spectrum of 38 Egyptian patients clinically suspected of having mitochondrial RC disorders
METHODS: A total of 38 patients (female, n=18 (47.4%); male, n=20 (52.6%)) clinically suspected of having mitochondrial diseases had been referred to the Inherited Metabolic Diseases Laboratory at Cairo University Children's Hospital. Laboratory investigations and analyses included histochemical staining of cytochrome c oxidase and succinate dehydrogenase in muscle biopsies, as well as spectrophotometric assays of RC complexes in muscle homogenates
RESULTS: Twenty-three patients (60.5%) were diagnosed with different RC enzyme deficiencies. Fifteen patients (65%) had complex I deficiency and all of them also had lactic acidosis (mean (standard deviation)) plasma lactate concentration of 4 (1.65) mmol/L). Two patients (9%) with marked complex IV deficiency both showed COX-negative ragged red fibers (RRFs) on histochemical staining. Combined complex I and complex II deficiency with scattered COX-stain deficiency and RRFs was diagnosed in 2 patients (5.25%), while a further 2 patients (5.26%) had combined (complex I, II+III, complex IV) deficiencies. Isolated complex II deficiency was diagnosed in 2 patients (5.26%) and 15 (39.5%) patients had normal RC enzyme activities
CONCLUSION: Biochemical assay of RC complexes is considered the cornerstone for diagnosis of RC complex mitochondrial disorders. These disorders are common among Egyptian paediatric patients
Mitochondrial disorders (MDs) are an expanding heterogeneous group of disorders caused by defects in intracellular energy production characterised by morphological and biochemical abnormalities of mitochondria that are usually associated with mitochondrial (mtDNA) mutations or nuclear gene defects.[1] They are one of the most common inborn errors of metabolism, with a conservative estimated prevalence of ~1: 5 000.[2]
The respiratory chain complexes (RCCs) consist of four enzymatic complexes (complexes I - IV) embedded in the inner mitochondrial membrane. These complexes catalyse the transfer of reducing equivalents from high-energy compounds (produced during the Krebs cycle) to oxygen, with the ultimate production of an electrochemical gradient through the inner mitochondrial membranes to drive the synthesis of adenosine triphosphate (ATP) by ATP synthase.[3]
Mitochondrial-associated myopathies are clinical features commonly affected in patients with a multisystem phenotype which includes chronic progressive external ophthalmoplegia, rhabdomyolysis, muscle fatigue and severe proximal weakness.[4]
Unfortunately, most paediatric patients present with nonspecific symptoms including developmental delay, failure to thrive, hypotonia, seizures and encephalopathy. This presents a diagnostic challenge to neuro-paediatricians and accounts for the considerable diagnostic delay.[5]
The diagnosis of MDs is usually based on diagnostic criteria that include clinical data, biochemical alterations (e.g. decreased RCC enzyme activity, and lactate acidosis), histological features, and genetic results (mtDNA or nuclear mitochondrial gene variants).[4]
Despite their invasiveness, biochemical assays measure the actual activity of individual oxidative phosphorylation (OXPHOS) enzyme (complexes I - V) in homogenised muscle tissues, and the results are normalised to the activity of a mitochondrial matrix enzyme, i.e. citrate synthase (CS).[1] The histochemical assessment of cytochrome c oxidase (complex IV, COX) and succinate dehydrogenase (complex II, succinate dehydrogenase (SDH)) activities assess RCC enzyme function in tissue biopsies, interrogating the activities of these complexes in the individual cell.[5] Complete molecular diagnosis requires several different methods for the detection and quantification of mtDNA gene variants and large deletions.[6]
Over the last 50 years, next-generation sequencing (NGS) technologies have accelerated our ability to detect molecular aetiologies of the disease, while whole-exome sequencing (WES) approaches have significantly improved the diagnosis of monogenic mitochondrial mimics and advanced our understanding of mitochondrial biology.[7]
A limited number of studies have focused on the diagnosis of MDs among Egyptian patients;[8-11] however, the role of the histochemical and the biochemical RCC assays for proper diagnosis of these disorders has not been clarified.
The current study focused on biochemical assays of RCCs and histochemical staining of muscle biopsies to elucidate their respective roles in the diagnosis of mitochondrial-related disorders in a sample of Egyptian paediatric patients.
Methods
The present study was conducted at the Inherited Metabolic Diseases Laboratory at Cairo University Children's Hospital (CUCH). Thirty-eight patients were included in the study after clinical recruitment based on diagnostic scoring for MDs[12] (Fig. 1). Thirty-eight age- and sex-matched controls were recruited Controls were attending the hospital for orthopaedic surgeries. Ethical approval was obtained from the Cairo University Research Ethical Committee (ref. no. N-15-2014). Parents of patients and controls gave written informed consent for all the procedures performed in the study. Written informed consent was obtained from each participant after a full explanation of the study protocol. All procedures included individual data were treated with confidentiality.
Routine laboratory investigations including liver function tests, kidney function tests, blood glucose, arterial blood gases, and plasma lactate were performed as first-line investigations to detect highly suspected cases. Amino acid and acyl carnitine profiles were done using liquid chromatography/mass spectrometry (LC/MS-MS) to exclude other inborn errors of metabolism that present with similar manifestations.
All patients were subjected to imaging studies, including brain magnetic resonance imaging (MRI), neurophysiological investigations, including ectroencephalogram (EEG) and electromyogram (EMG). Nerve conduction studies (NCS) and visual-evoked potentials (VEPs) were done for all patients. Some patients were subjected to a fundus examination.
Muscle biopsies from quadriceps muscles were performed under general anaesthesia for selected patients. For histochemical examinations, fresh transverse sections of muscle fibers from muscle biopsies were stained with haematoxylin and eosin, Gömöri stain, cytochrome c oxidase (COX) and succinate dehydrogenase (SDH), using staining kits (Bio-Optica S.p.A, Italy). Some muscle fibers were fixed in glutaraldehyde and routinely processed for electron microscopic examination. The rest of the biopsy was immediately preserved at -80° C for biochemical assays. All these procedures were performed for the control group.
For the biochemical RCC assay, biopsies were homogenised on ice using a conical tissue grinder in a 1:9 (w/v) ratio in potassium phosphate buffer pH 7.5 containing 0.25 M sucrose, 2 mmol ethylene diamine tetra acetic acid (EDTA) and 50 IU/mL heparin.[4]
Mitochondrial RCCs and CS enzymatic activities were determined by a spectrophotometric enzyme assay. Complex I (NADH: ubiquinone reductase; EC1.6.5.3) activity was measured by the rotenone-sensitive oxidation of NADH at 340 nm. Complex II/ III (succinate: cytochrome c reductase; EC1.3.5.1 and EC1.10.2.2) activity was measured by antimycin A-sensitive succinate-dependent reduction of cytochrome c at 550 nm. Complex IV (cytochrome c oxidase; EC1.9.3.1) activity was measured by the potassium-cyanide-sensitive oxidation of reduced cytochrome c at 550 nm. CS (EC2.3.3.1) activity was determined by the formation of 5-to-2-nitrobenzoic acid following the incubation of tissue homogenate with acetyl-CoA, oxaloacetate, and 5,5'-dithio-bis-(2-nitrobenzoic acid) at 412 nm using UV/visible spectrophotometry (Ultra spec 2100 pro; Amersham, UK) supported by Swift-II software (version 2.06; Fisher Scientific, UK).[4]
Statistical analyses
Data were analysed using SPSS advanced statistics version 20 (IBM Corp., USA). Qualitative data were expressed using numbers and frequencies. Quantitative data were expressed using mean (standard deviation (SD)) or median (range) as appropriate and categorical variables were expressed as percentages. All mitochondrial enzymes were expressed as a ratio to the activity of CS and complex II as an internal mitochondrial marker enzyme to standardise the mitochondrial enrichment of the sample. Activities were expressed in nmol/mL/mg protein and the range, mean (SD) and Z-scores were calculated for each RCC enzyme activity. Statistical significance between patients' results and controls was expressed as the p-value (significance considered if p<0.0001).
Results
The present study included 38 paediatric patients (female, n=18 (47.4%); male, n=20 (52.6%)) and 38 controls (female, n=24 (63%); male, n=16 (37%)). The median (range) age of patients and controls was 6(1 - 15) and 4.5 (1 - 16) years, respectively.
Positive consanguinity was remarkable in 84% patients (n=32) and negative consanguinity in 16% (n=6). The demographic data and the clinical presentations of the patients and controls are described in Table 1.
Atrophic brain changes were observed in the brain MRI of 10 cases (26.3%): 4 had normal RC enzyme activity (n=4/10; 40%), while the remaining 6 (60%) cases had variable degrees and types of enzyme deficiencies. Bilateral and symmetrical abnormal signal intensity in the basal ganglia was the main finding in 7 cases (18.4%) and all showed RCC enzyme deficiency. Bilateral white matter demyelination was seen in 5 cases (39.5%) of whom all had complex I deficiency, while bilateral cerebellar demyelination was seen in 4 patients (10.5%), with RCC enzyme deficiency in 3 patients (7.9%). Normal brain imaging was observed in 12 patients (32.6%). Radiological and neuroimaging findings of the patients are shown in Table 2.

Electron microscopic examination showed accumulation of sub-sarcolemmal aggregates of mitochondria in 14 patients (36.8 %), and biochemical RCC enzyme activity assays verified 8 out of 14 patients with complex I deficiency (57.2%): 2 patients had isolated complex II deficiency (14.3%); 2 had combined complex I, II+III, and IV deficiency (14.3%); 1 patient had isolated complex IV deficiency (7.1%); and 1 patient had normal enzyme activity (7.1%).
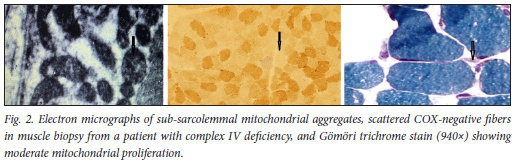
Histochemical investigations showed COX-negative ragged red fibers (RRFs) in 8 patients (21.1%), scattered COX-stain deficiency with RRF in 4 patients (10.5%), atrophied muscle changes with RRFs in 6 patients (15.8%) (Fig. 2), nonspecific muscle changes in 5 patients (13.2%) and a normal pattern in 15 patients (39.5%) (Table 3).
Biochemical measurement of RCC enzymes in muscle homogenates confirmed enzyme deficiency in 23 patients (60.5%) when compared with citrate synthase and complex II enzyme activities. Fifteen patients (65%) showed complex I deficiency (Fig. 3). All of the patients with complex I deficiency had lactic acidosis (mean (SD) plasma lactate 2.8 (1.18) mmol/L). Complex IV enzyme activity assays verified 2 cases (9%) (Fig. 3). Both patients presented with convulsions, epileptogenic focus on EEG, and Cox-stain deficient RRFs on histopathological examination. Two patients (9%) had combined complex I and complex II deficiencies, two patients (9%) had combined (complex I, II+III, complex IV) deficiencies, two patients (9%) (Fig. 3) had isolated complex II deficiency, and 15 patients had normal RCC enzyme activities (39.5%). Tables 3 and 4 show a summary of the results of the histochemical and biochemical analyses. The supplementary table (https://www.samedical.org/file/1956) summarises demographic, clinical, radiological, biochemical and histopathological data of the patients (N=38).
Discussion
Many Egyptian studies have discussed MDs from the clinical, radiological and pathological points without verifying the role of biochemical mitochondrial enzyme activity measurements as a step for determining isolated and combined complex deficiencies in individuals.
El-Bassyouni et al.[14] illustrated the clinical pathological, and biochemical diagnosis of mitochondrial diseases in 19 Egyptian patients, in whom they only measured plasma lactate, lactate/pyruvate ratio, and serum cytochrome c using an enzyme-linked immunoassay (ELISA).[14] Selim et al
[8] performed their studies on 31 patients to confirm the role of brain MRI and MRS as non-invasive diagnostic procedures,[8] while El-Ettribi et al.[9] investigated the most common mtDNA point mutations based on clinical suspicion in only 8 Egyptian patients.[9] Selim et al.[10] discussed the role of clinical, pathological, biochemical and molecular diagnosis of mitochondrial neurogastrointestinal encephalopathy in 3 Egyptian patients.[10]
Diagnosing mitochondrial disorders remains a major challenge for the clinician owing to variable age at onset, clinical presentations and their genetic heterogeneity. The diagnosis often remains uncertain until muscle biopsy investigations are conducted and genetic tests confirm an abnormality. [15] Walker et al.[16] were the first to establish a set of major and minor criteria for the identification of MDs.[16] The Walker criteria were then modified by Bernier et al.,[17] and are now known as the modified Walker criteria.[17] Other attempts to define diagnostic criteria included the Nijmegen Centre for Mitochondrial Disorders scoring system and the Mitochondrial Disease Criteria.
In the present study, lactic acidosis was the most common finding (n=28 (73.7%)). Fatal infantile lactic acidosis (FILA) has been reported most frequently in children with complex I deficiency of the mitochondrial respiratory chain disorder.[18]
Convulsions were the second most frequent clinical presentation representing 31.6% (n=12) of patients. Lee et al.[2l] performed a study of 125 patients clinically suspected of having Leigh syndrome. Among the patients, 25 were identified as having mitochondrial DNA associated with Leigh syndrome, while 14 out of the 25 (56%) presented with seizures.[21]
As Egypt is an African country, it was crucial to compare our findings with those of a study of African patients by Smuts et al.,[22] who reported that myopathy was the most frequent clinical presentation (62.5%), followed by encephalomyopathy (30%) and encephalopathy (7.5%), while encephalomyopathy was the predominant finding (60%) among the white patients, followed by an equal frequency of encephalopathy and myopathy (20%).[22]
In contrast, an audit of all mitochondrial disease genetic tests (N=1614) performed in Cape Town, South Africa (SA), by Meldau et al[23] found that muscle phenotypes are not commonly encountered in SA paediatric patients.[23]
This variation may be due to the difference in the examined sample size, with more conclusive results reported by Meldau et al.[23]
Histopathological findings matched biochemical assay results of mitochondrial enzymes, as 10 patients (66.7%) showed neurogenic muscle atrophy in muscle biopsies, with accumulation of sub-sarcolemmal aggregates of mitochondria visible in electron micrographs. Rollins et al.[24] evaluated 118 muscle biopsy specimens from patients of all ages with mitochondrial diseases and reported normal findings on light microscopy of 45% of the specimens, RRFs in 2.5% and cytochrome c oxidase-negative fibers were found in 7% (53).[24] A previous Egyptian study reported RRFs on modified Gömöri trichrome stain of muscle biopsies from all patients in the study as an associative finding with MCDs.[1l] Selim et al. [10] also illustrated the importance of histochemistry in muscle biopsies obtained from suspected MD patients in increasing Morava scoring for clinical diagnosis.[10]
The present study verified complex I deficiency in 65% (n=15/23) of patients with RCC enzyme deficiency, a finding matching previously published studies in mitochondrial diseases.[10,23,24] In our study, all patients with complex I deficiency had lactic acidosis, but developmental delay was notable in only 6 patients (n=6/15; 40%), while RRFs were the main pathological finding in 9 patients (n=9/15; 60%). Owing to the clinical heterogeneity of complex I disorders, patients often do not present with the traditional hallmarks of mitochondrial disease. This reinforces the importance of biochemical and metabolic screening as appropriate methods to diagnose complex I deficiency in paediatric patients.[24]
Mitochondrial complex II deficiency is an autosomal-recessive disorder with a highly variable phenotype. It is exclusively nuclear-encoded with some patients having multisystem involvement of the brain, heart, muscles, liver and kidneys. This may result in death in infancy, whereas some patients have only isolated cardiac or muscle involvement with onset in adulthood and normal cognition.[2] In the present study, 2 patients had isolated complex II deficiency (n=2/23, 8.7%), both with positive consanguinity: one presented with lactic acidosis and bulbar palsy with abnormal basal ganglia signals in the brain MRI, while the other presented with exercise intolerance, ptosis, and cardiomyopathy.
Isolated complex IV deficiency was notable in 2 patients (n=2/23; 8.7%). One patient with positive consanguinity presented with convulsions, peripheral neuropathy and hypertrophic cardiomyopathy, with white matter demyelination visible on brain MRI, as well as COX-stain deficiency and RRFs on histopathological investigation. The other patient presented with delayed motor development, lactic acidosis, convulsions, abnormal basal ganglia signals on brain MRI, and COX-stain deficiency with RRFs and sub-sarcolemmal aggregates on histopathology. Fatal infantile cardio-encephalomyopathy with COX-stain deficiency was the main finding in a previous study which determined that clinical phenotype caused by mutations in the human SC02 assembly gene causing complex IV deficiency presented mainly with Leigh syndrome.[25]
Although isolated complex deficiency was notable in this study, combined complex deficiency was a remarkable finding: 2 patients (n=2/23; 8.7%) had combined complex I and II deficiency; and 2 patients (n=2/23; 8.7%) had complex I, II, III and IV deficiency. On the other hand, in their study of 191 subjects, Smuts et al.[22]
(including both African and white patients) illustrated that single-enzyme deficiencies were more common than combined deficiencies in white patients (60.0%), but combined deficiencies were found in 67.5% of African patients.[22]
Numerous co-factors also play an essential role in mitochondrial energy metabolism and some of these cofactors are required for the function of RCC enzymes, such as coenzyme Q, iron-sulphur clusters, riboflavin and haem - deficiencies typically result in defects of more than one respiratory enzyme. [20]
The lack of genetic data limited the present study but important logistical and procedural challenges have been overcome, in addition to using control samples for refinement of analyses and reference values.
Conclusion
The results of the present study confirm the clinical heterogeneity of mitochondrial diseases and the importance of biochemical assays of RCC enzymes in muscle biopsies in suspected patients. Histopathological investigations of muscle biopsies will raise the confirmatory diagnosis (Morava score) and aids biochemical diagnosis.
Despite the limited sample size, the presence of 60.5% of patients with respiratory chain enzyme defects confirms that mitochondrial diseases are not rare among Egyptian paediatric patients and there is a need for proper selection of patients based on strict inclusion criteria. Histochemical, histopathological and biochemical respiratory chain enzymes assay in muscle biopsy are needed for better diagnosis and treatment of mitochondrial diseases. Molecular analysis of respiratory chain complex deficiencies is crucial for the elucidation of the underlying genetic basis of such disorders in Egyptian patients. National programmess are needed for early detection, intervention and management of such disorders in the Egyptian population.
Declaration. None.
Acknowledgements. We thank the patients, controls and their families for participation in the study, as well as Prof. Dr Fayza Hassan, Professor of Chemical Pathology in the Faculty of Medicine; Dr Mohammed Mostafa Allak in the Department of Pediatrics, Faculty of Medicine, for collecting the clinical data from the patients' samples for analysis at Gent University; Dr Abdel Bary of the Surgery Department, Faculty of Medicine, Cairo University, the surgeon who collected the muscle biopsies to do the biochemical/histochemical assays; and Prof Sawsan Hassan, Professor of Pediatrics, Faculty of Medicine, Cairo University, who supported the approvals for the fund used for purchasing the equipment needed for the assay.
Author contributions. LAS was the principal investigator and was responsible for the study design. RV and IS managed the practical work protocol and interpretation of results. DMA was responsible for sample collection, performing the practical work according to the protocol data analysis, manuscript writing and submission. GAN was responsible for histopathological examinations and revision of results. DAM was responsible for manuscript editing and reviewing, revision of clinical data and biochemical assays conducted at Gent University, as well as interpretation of results.
Funding. The histochemical and biochemical assays in Egypt were supported by the Cairo University research fund. The biochemical assays at Gent University in Belgium were supported by a grant from the Fund for Scientific Research Belgium (grant no. G.0666.06).
Conflicts of interest. None.
References
1. Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc2012;7(6):1235-1246. https://doi.org/10.1038/nprot.2012.058 [ Links ]
2. Gorman GS, Schaefer AM, Ng Y, et al. Prevalence of nuclear and mitochondria] DNA mutations related to adult mitochondrial disease. Ann Neuro 2015;77(5):753-759. https://doi.org/10.1002/ana.24362 [ Links ]
3. Smeitink JA, Zeviani M, Turnbull DM, Jacobs HT. Mitochondrial medicine: A metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metab2006;3(1):9-13. https://doi.Org/10.1016/j.cmet.2005.12.001 [ Links ]
4. Chretien D, Rustin P, Bourgeron T, Rotig A, Saudubray JM, Munnich A. Reference charts for respiratory chain activities in human tissues. Clin Chim Acta 1994;228(1):53-70. https://doi.org/10.1016/0009-8981(94)90056-6 [ Links ]
5. Yarham JW, Elson JL, Blakely EL, Mcfarland R, Taylor RW Mitochondrial tRNA mutations and disease. Wiley Interdiscip RevRNA2010;1(2):304-324. https://doi.org/10.1002/wrna.27 [ Links ]
6. Frazier AE, Thorburn DR, Compton AG. Mitochondrial energy generation disorders: Genes, mechanisms, and clues to pathology. J Biol Chem 2019;294(14):5386-5895. https://doi.org/10.1074/jbc.R117.809194 [ Links ]
7. Sulaiman SA, Rani ZM, Radin FZM, M NAA. Advancement in the diagnosis of mitochondrial diseases. J Transi Gen Genomics 2020;4:159-187. https://doi.org/10.20517/jtgg.2020.27 [ Links ]
8. Selim LA, Ghaffar HA, Hamdi MM, Hagras AM, Nakhla GA, Kamel AM. A clinicopathological and brain MRI study in Egyptian children with mitochondrial disorders. J Pediatr Neurol 2005;3(4):207-215. https://doi.org/10.1055/s-0035-1557283 [ Links ]
9. Al-Ettribi GMM, Effat LK, El-Bassyouni HT, Zaki MS, Shanab G, Karim AM. Screening seven common mitochondrial mutations in 28 Egyptian patients with suspected mitochondrial disease. Middle East J Med Genet 2013;2(1):28-37. https://doi.org/10.1097/01.MXE.0000422779.05483.d7 [ Links ]
10. Selim L, van Coster R, Mehaney D, et al. Mitochondrial neurogastrointestinal encephalopathy: Clinical, biochemical and molecular study in three Egyptian patients. Genet Couns 2016;27(2): 193-205. [ Links ]
11. Al-Ettribi GMM, Effat LK, El-Bassyouni HT, Zaki MS, Shanab G, Karim AM. Clinical and molecular findings in eight Egyptian patients with suspected mitochondrial disorders and optic atrophy. Egypt J Med Hum Genet 2013;14(1):37-47. https://doi.Org/10.1016/j.ejmhg.2012.08.002 [ Links ]
12. Morava E, van den Heuvel L, Hoi F, et al. Mitochondrial disease criteria: Diagnostic applications in children. Neurology 2006;67(10):1823-1826. https://doi.org/10.1212/01.wnl.0000244435.27645.54 [ Links ]
13. Thorburn DR. Practical problems in detecting abnormal mitochondrial function and genomes. Hum Reprod 2000;2(15):57-67. https://doi.org/10.1093/humrep/15.suppl_2.57 [ Links ]
14. El-Bassyouni HT, Abdallah HA, Shousha AS, Hassan Ibraheim WHI. Clinical and biochemical aspects of mitochondrial disorders in Egyptian patients. Int J Child Neuropsychiatry 2004;14(1):41-50. [ Links ]
15. Liang C, Ahmad K, Sue CM. The broadening spectrum of mitochondrial disease: Shifts in the diagnostic paradigm. Biochim Biophys Acta 2014;1840(4):1360-1367. https://doi.org/10.1016/j.bbagen.2013 [ Links ]
16. Scaglia F, Towbin JA, Craigen WJ, et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics 2004;114(4):925-931. https://doi.org/10.1542/peds.2004-0718 [ Links ]
17. Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, Thorburn DR. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 2002;59(9):1406-1411. https://doi.org/10.1212/01.wnl.0000033795.17156.00 [ Links ]
18. Parikh S, Goldstein A, Koenig MK, et al. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet Med 2015;17(9):689-701. https://doi.org/10.1038/gim.2014.177 [ Links ]
19. Pronicka E, Piekutowska-Abramczuk D, Ciara E, et al. New perspective in diagnostics of mitochondrial disorders: Two years' experience with whole-exome sequencing at a national paediatric centre. J Transi Med 2016;14(1):174. https://doi.org/10.1186/sl2967-016-0930-9 [ Links ]
20. Fassone E, Rahman S. Complex I deficiency: Clinical features, biochemistry and molecular genetics. J Med Genet 2012;49(9):578-590. https://doi.org/10.1136/jmedgenet-2012-101159 [ Links ]
21. Lee S, Na JH, Lee YM. Epilepsy in Leigh syndrome with mitochondrial DNA mutations. Front Neurol 2019;10:496. https://doi.org/10.3389/fneur.2019.00496 [ Links ]
22. Smuts I, Louw R, Du Toit H, Klopper B, Mienie LJ, Van Der Westhuizen FH. An overview of a cohort of South African patients with mitochondrial disorders. J Inherit Metab Dis 2010;33(Suppl 3):S95-S104. https://doi.org/10.1007/sl0545-0099031-8 [ Links ]
23. Meldau S, Owen PE, Khan K, Riordan TG. Mitochondrial molecular genetic results in a South African cohort: Divergent mitochondrial and nuclear DNA findings. J Clin Pathol 2022;75:34-38. https://doi.org/10.1136/jclinpath-2020-207026 [ Links ]
24. Rollins S, Prayson RA, McMahon JT, Cohen BH. Diagnostic yield muscle biopsy in patients with clinical evidence of mitochondrial cytopathy. Am J Clin Pathol 2001;116(3):326-330. https://doi.org/10.1309/WATB-W4QV-NA53-B9MY [ Links ]
25. Sánchez-Caballero L, Ruzzenente B, Bianchi L, et al. Mutations in complex I assembly factor TMEM126B result in muscle weakness and isolated complex I deficiency. Am J Hum Genet 2016;99(1):208-216. https://doi.org/10.1016/j.ajhg.2016.05.022 [ Links ]
 Correspondence:
Correspondence:
D M Abdou
doaa.mohamed@kasralainy.edu.eg
Accepted 6 June 2022


{kind=link}
{kind=link}