










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Child Health
versión On-line ISSN 1999-7671
versión impresa ISSN 1994-3032
S. Afr. j. child health vol.17 no.1 Pretoria 2023
http://dx.doi.org/10.7196/SAJCH.2023.v17i1.1942
RESEARCH
A review of spinal muscular atrophy in black South African paediatric patients
K FlackI; M P K HauptfleischII; L G ScherIII
IFCPaed (SA); Department of Paediatrics, University of the Witwatersrand, Johannesburg, South Africa
IICert Paed Neuro (SA); Division of Paediatric Neurology, Department of Paediatrics, Chris Hani Baragwanath Academic Hospital, University of the Witwatersrand, Johannesburg, South Africa
IIIFCPaed (SA); Division of Paediatric Neurology, Department of Paediatrics, Charlotte Maxeke Johannesburg Academic Hospital, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
BACKGROUND: Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disorder that is present in all populations and results in muscle weakness owing to anterior horn cell degeneration. SMA is divided into three clinical subtypes and is an important genetic cause of morbidity and mortality but has not been well studied in sub-Saharan Africa
OBJECTIVE: This study aims to describe the clinical features and genetic findings in black patients with SMA presenting to the Division of Paediatric Neurology at Chris Hani Baragwanath Academic Hospital (CHBAH) over a 30-year period
METHOD: This study was a retrospective review of patient records. The study population was black paediatric neurology patients with clinical SMA, who attended CHBAH Neurology Clinic between 1988 and 2018. Patients were categorised into SMA type 1, 2 or 3 based on their neurology assessment and clinical features were recorded
RESULTS: The clinical findings in the study population (with SMA), i.e. hypotonia, areflexia and tongue fasciculations, were similar to those found in international studies. More than half of the patients (65.6%; n=86/131) had genetic tests, of which 84.8% were positive for SMA. This value was significantly higher than previously reported results from South Africa. At least 23.6% (n=31/131) had facial involvement
CONCLUSIONS: This study adds to the limited body of research on SMA in sub-Saharan Africa and highlights the lower frequency of a homozygous deletion seen in the black South African population compared with the expected 95% worldwide
Keywords: Spinal muscular atrophy; SMA; neurology; survival motor neuron protein; SMN; genetics.
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disorder, which results in progressive and symmetrical muscle weakness owing to irreversible anterior horn cell degeneration.[1] The incidence of SMA in the USA is estimated at 1 in 6 000 to 1 in 11 000 live births, with an estimated 1:38 carrier frequency,[2] thus making it the second most common neuromuscular disorder after Duchenne muscular dystrophy.[3] SMA is present in all populations but Verhaart et al.[4] suggested a slightly lower incidence in patients of sub-Saharan ancestry of 1 in 18 808. However, a South African (SA) study suggested that SMA is more common than previously thought, with an incidence of 1 in 3 574 in the black SA population and 1 in 1 945 in those with European ancestry.[3]
SMA is divided into three clinical subtypes that represent a spectrum of clinical severity. The clinical picture seen in SMA is due to anterior horn cell degeneration in the spinal cord, leading to muscle weakness and associated complications. Type 1 SMA usually presents before 6 months of age and patients never sit unsupported.[5] It is the most common type, affecting up to 50% of all SMA patients. Type 1 SMA patients usually develop respiratory failure within the first 2 years of life and less than 65% survive beyond this age.[3] The outcome is dependent on respiratory involvement.
Type 2 SMA or 'sitters' achieve the gross motor milestone of sitting unsupported but are unable to stand or walk. The age of onset is usually between 6 and 12 months. This intermediate form of SMA is generally slower to progress, with a variable prognosis.[5] Most children with type 2 SMA survive beyond the age of 5 years and survival thereafter is dependent on the development of complications involving the respiratory system.[3]
Type 3 SMA describes a group clinically defined as 'walkers'. The onset of observable symptoms in this group is usually after age 12 to 18 months. This is considered a mild, chronic form of SMA with little effect on patient lifespan. These patients can walk but over time develop proximal weakness involving the lower limbs more than the upper limbs.[6] These patients have a normal life expectancy.
Two extremes of these subtypes have also been described, type 0 SMA and type 4 SMA which were not included in the present study. Type 0 SMA is a rare form which develops in utero[7] and is usually fatal by age 6 months.[8] Type 4 SMA is the mildest form of the disease, with onset of symptoms in adulthood and usually in the third decade of life.
SMA is a leading genetic cause of infant mortality[7] with an autosomal recessive inheritance pattern caused by a homozygous deletion or mutation of the survival motor neuron (SMN1) gene on chromosome 5q13 in 95% of patients worldwide.[3] However, SA studies have proposed a different genetic cause with only 51% of SA black patients having the homozygous deletion of the SMN1 gene. One such study was conducted at Chris Hani Baragwanath Academic Hospital (CHBAH).[3] Another study conducted in Western Cape Province concluded that the genetic findings in their patients were in keeping with those that are internationally expected. This group included only 12 black SA patients and was therefore much smaller than the CHBAH study sample size.[8]
Autosomal-recessive SMA is the most common form, but X-linked and autosomal-dominant cases have been described.[9] The genetic defect was identified in 1995 and a wide range of clinical severity was noted. [10] It was discovered that 2 forms of the SMN gene exist, i.e. SMN1 and SMN2. SMN2 and SMN1 are identical, except for a single nucleotide change at exon 7 (of SMN2). The resulting protein produced by transcription at SMN2 is shorter, less functional and degrades faster than the protein produced at SMN1. Only ~10% of the protein produced by the SMN2 gene is fully functional. As patients with SMA have a deletion of SMN1, they rely on the SMN protein produced at SMN2 for survival. A correlation between disease severity and SMN2 copy number has been found, i.e. the more SMN2 copies a person has, the less severe the clinical phenotype of SMA.[11]
The aim of the present study was to describe the clinical features and genetic findings in black SA patients with SMA presenting to the Division of Paediatric Neurology at CHBAH over a 30-year period and compare results with two previous studies from this site as well as other SA publications. Black SA patients were the focus of the study, as previous studies suggested there could be a different genetic basis for SMA in this group.[3] There are no studies with a comparable size available and there is an overall paucity of data from sub-Saharan Africa. We know that other diseases have been found to have different disease manifestations in black Africans as a result of a different mutation than that seen in the white population, e.g. cystic fibrosis, galactosaemia and Fanconi's anaemia.[3,10] This provides the rationale for the study.
Methods
CHBAH is the only large public hospital in Soweto, SA, and the third largest in the world. The paediatric neurology clinic receives referrals from the surrounding area and the greater Gauteng Province, as well as other provinces and southern African countries where specialist expertise is lacking.
The present study was a retrospective review of patient records captured by the paediatric neurology team at CHBAH during the period 1988 - 2018. All the files of patients with a diagnosis of SMA were reviewed and the study population included all black paediatric neurology patients with clinical, genetic and/or histological diagnosis of SMA who attended CHBAH Neurology Clinic during this 30-year period.
All patients presenting to the Division of Paediatric Neurology at CHBAH with clinical features in keeping with the diagnosis of SMA type 1, 2 or 3 (as determined by the paediatric neurologists) were included. This group also included all patients presenting with a confirmed genetic result or with a confirmed muscle biopsy result.
Genetic testing for the homozygous SMN1 deletion became available at the National Health Laboratory Service in 1996. The standard test uses polymerase chain reaction (PCR) and restriction enzyme analysis. Patients who presented prior to 1996 were subjected to muscle biopsy, which typically shows large groups of atrophic fibres interspersed with hypertrophied and normal fibres.
A small number of these biopsy patients later had genetic testing done as part of a study. Multiplex ligation-dependent probe amplification (MLPA) testing is not done as a routine diagnostic assay for SMA but has been used in research projects to assess heterozygous deletions and SMN copy numbers. Patients for whom the diagnosis of SMA was in doubt were excluded.
Data were entered into a confidential and secure Microsoft Excel (Microsoft Corp., USA) spreadsheet. Each patient file was reviewed and multiple variables were entered for each patient. The age at first presentation to the neurology clinic was captured and the age of onset based on history was recorded as either less than 6 months, 6 to 18 months or greater than 18 months. The grade of SMA at diagnosis, i.e. type 1, 2 or 3, was determined from the initial neurology assessment and the clinical features present at the first assessment were captured. Files were further reviewed for genetic or muscle biopsy results.
The resulting spreadsheet was exported into STATA for analysis. Comparative tables were generated and ^-values were determined using either Fischer's exact test or the chi-square test.
Ethics approval
This study was approved unconditionally by the Human Research Ethics Committee of the University of the Witwatersrand (ref. no. M190317).
Results
A total of 145 patients with SMA were assessed at CHBAH from 1988 - 2018. As the focus of the present study was SMA in black SA patients, 14 patients of white, Asian or mixed ancestry were excluded. A total of 131 patient files were subsequently analysed.
The patient files contained information on clinical findings at initial presentation to the neurology clinic and biopsy or genetic results. Only 45 patient files also contained information on progression of clinical features and therefore only clinical features at the first visit were reviewed in the present study.
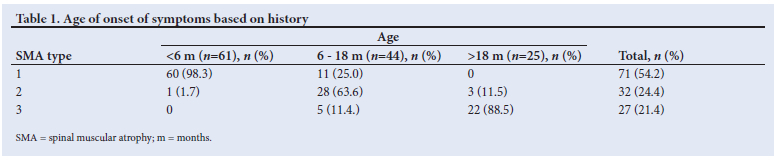
The median (IQR) age of presentation of all SMA patients to CHBAH paediatric neurology clinic was 15 (6 - 34) months. The median (IQR) age at presentation of patients was 7 (4 - 11) months for type 1 SMA, 24 (16 - 32) months for type 2 SMA and 56 (36 - 72) months for type 3.
Paediatric neurologists working at CHBAH made the diagnosis of SMA clinically and later confirmed the diagnosis with muscle biopsy (prior to the availability of genetic testing) or genetic testing. SMA affected males and females equally, with 51.2% males and 48.8% females in our study group. The most common complaint from parents in infants was weakness or a floppy child in infancy. SMA type 3 cases would present with a history of frequent falling or difficulty walking. Most of the SMA type 1 (84.5%) patients gave a history of symptoms starting before the age of 6 months. Similarly, the majority of SMA type 2 (87.5%) patients had onset of symptoms between age 6 and 18 months. Most of the type 3 SMA (81.4%) patients developed symptoms after the age of 18 months.
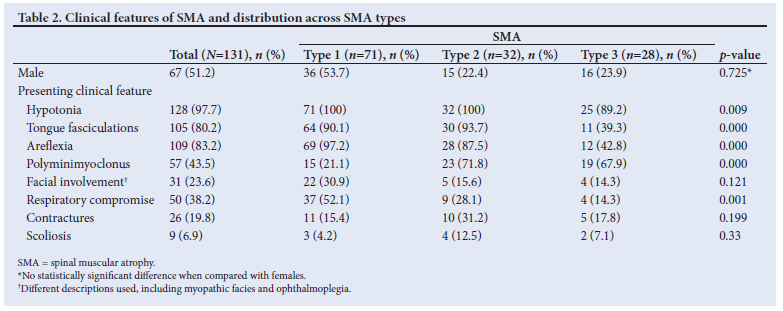
Table 2 shows the presenting clinical features and gender across all SMA types.
The primary clinical features documented were hypotonia (97.7%), absent or diminished reflexes (83.2%), and tongue fasciculation (80.2%).
Hypotonia was present in 100% of type 1 and type 2 patients and in 89% of the type 3 patients. Absent lower-limb reflexes were found in 98% of patients, 22 patients (16.8%) had preserved upper limb reflexes and only 2 patients (1.5%) had all reflexes elicited at presentation. Both of these patients were type 3 SMA, with one having a homozygous deletion of SMN1 and the other having a negative genetic result but positive biopsy result. More than 90% of type 1 SMA patients had fasciculation of the tongue while fasciculations were only present in 39% of type 3 SMA patients. Further analysis showed that 36.6% (n=48) of patients had both tongue fasiculations and polyminimyoclonus, 9.9% (n=13) had polyminimyoclonus only, and 46.5% (n=61) had tongue fasciculations only.
Closer review of our data showed 23.6% («=31) of 131 patients had facial involvement, myopathic facies with or without ophthalmoplegia. Of these, 35.4% (n=11) had positive genetic results and 48.3% (n=15) had positive muscle biopsy results confirming the diagnosis of SMA. Some patients had already developed complications of the disease at the time of presentation. The presence of contractures, scoliosis and respiratory compromise was also reviewed. Thirty-seven percent of the patients had respiratory compromise at presentation. Scoliosis and contractures were the least common findings.
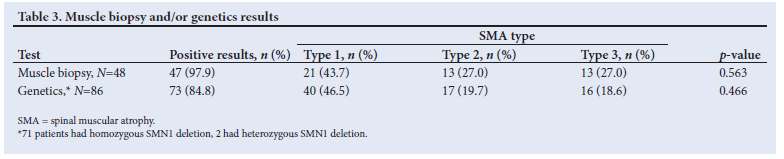
Muscle biopsy and genetic results are shown in Table 3. Most of the patients in the study population had either a muscle biopsy or genetic testing done. Nearly all the muscle biopsies (97.9%; n=48) were positive. The only patient with a negative biopsy showed nonspecific histological changes but had a confirmed homozygous deletion of the SMN1 gene. This patient presented with SMA type 3. Of the patients who had genetic testing (n=86), 82.5% (n=71/86) had a homozygous deletion and 2.3% (n=2/86) had a heterozygous deletion. These patients had a single deletion of SMN1 and a presumed mutation not detected by traditional genetic testing. Both of these patients had positive muscle biopsies.
A total of 17 patients had both genetic testing and muscle biopsy; 5 patients had a biopsy performed after genetic testing and of these patients, 2 had heterozygous deletions, 2 had no deletions and 1 result was inconclusive. All 5 of the biopsies done confirmed SMA.
A total of 5 patients had no biopsy or genetic testing; 4 of these patients had planned biopsies but were lost to follow-up and I patient's family refused a biopsy. All 5 of these patients were seen before genetic testing was available and SMA diagnosis was made on clinical presentation only.
Discussion
SMA is an important genetic cause of infant mortality [1] and has not been well studied in sub-Saharan Africa.[14] In the present study, males and females were equally affected and the median (IQR) age of presentation across all types of SMA was 15 (6 - 34) months. SMA type 1 is the most common form of the disease (54.2%), with patients typically presenting by 7 months of age and onset of symptoms occurring before 6 months of age in 98.3%. The results of the present study are similar to previously published international data.[5] Although the majority of type 2 SMA patients had onset of symptoms between 6 and 18 months of age, most only presented at around 24 months of age. Delay in presentation is common in the SA public healthcare system, owing to multiple factors including access to healthcare, cultural beliefs and level of patient education.
The present study focused on a single racial group, as previous studies from SA suggested that there may be a different genetic basis for SMA in black South Africans.[3] The main finding was that 84.8% (n=73/86) of the genetic results for patients were positive, with the majority having a homozygous deletion of the SMN1 gene (82.5%). This result is significantly higher than what had previously been reported in a study at CHBAH where only 51% of the patients had the expected SMN1 deletion.[3] This result may be attributed to more widespread access to genetic testing over the last two decades.
Our study also differs from what has been reported at Red Cross War Memorial Children's Hospital in Cape Town.[10] The present study featured a large sample size of 131 black patients, compared with 12 black patients in the Cape Town study, which also strictly excluded all patients with facial involvement.[10] Our study findings suggest that excluding these patients results in up to 1 in 4 patients (23.6%) with confirmed SMA potentially being missed. A study in KwaZulu-Natal[15] showed facial involvement in 80% of type 1 SMA patients, with 73% having bulbar weakness. This study was done prior to the availability of genetic testing and therefore alternate diagnoses could be considered.
Prior to the availability of genetic testing, muscle biopsy was the mainstay of diagnosis. Almost all patients with available muscle biopsy results (97.9%) had histologically confirmed diagnoses of SMA, which had a strong correlation with clinical diagnosis. Despite the fact there is a strong correlation between histological and clinical diagnosis, muscle biopsy is not the preferred initial test for SMA for at least two reasons: (i) the procedure is invasive and painful for patients;[12] and (ii) there are additional associated risks, e.g. general anaesthetic use in a patient who may already have respiratory compromise. Muscle biopsy cannot provide a genetic profile to assist with further genetic counselling for parents and patients, therefore genetic testing remains the preferred first-line test. In patients where there is a strong clinical suspicion of SMA despite a negative genetic test and inconclusive muscle biopsy results, MLPA is available as an option to look for alternate diagnoses.
A recent publication[15] showed that MLPA is not a suitable test to determine carrier status in black South Africans, as up to 50% have multiple SMN1 copy numbers. This has the potential to mask a heterozygous deletion and will not detect carrier status in these cases. The same study reported only 8.3% of the clinically suggestive patients had a heterozygous deletion, which was lower than previously reported but higher than what was found in our study.[3,16]
As genetic testing evolves and gene-based therapies become available, a further understanding of the genetic basis for disease in black South Africans is needed.
Clinical diagnosis is the first step and early intervention as a multidisciplinary team, including paediatric neurologists and rehabilitation specialists, may improve quality of life for these patients.
Study limitations
Owing to the retrospective nature of the study, medical records compiled by multiple clinicians were reviewed. Furthermore, it was carried out at a single centre and included only one ethnic group. A review of results at other sites with comparable patient volumes may be warranted.
Conclusion
The present study adds to the limited body of research on SMA in sub-Saharan Africa. The main finding from this study, that 82.5% of the genetic results for patients were homozygous-deletion positive, is significantly higher than what had previously been reported in SA. This is still lower than what has been reported internationally and lower than what is found in white patients. We found that the clinical presentation of the present study population was similar to what had been reported internationally. Facial involvement is an important clinical sign noted in up to 25% of patients. The focus on a single racial group was justified, as previous SA studies suggested that there may be a different genetic basis for SMA in black South Africans. Ongoing research is needed in this area.
Declaration. This manuscript was submitted in partifal fulfilment of the requirements for KF's MMed (Paed).
Acknowledgements. The authors wish to thank the Paediatric Neurology Department at CHBAH, as well as Elana Vorster and Fahmida Essop from the Division of Human Genetics.
Author contributions. KF: literature review, data collection and analysis. KF, MH, LGS: wrote and edited the manuscript.
Funding. None.
Conflicts of interest. None.
References
1. Markowitz JA, Singh P, Darras BT. Spinal muscular atrophy: A clinical and research update. Pediatr Neurol 2012;46(1):1-12. https://doi.org/10.1016/j.pediatrneurol.2011.09.001 [ Links ]
2. Lin CW, Kalb SJ, Yeh WS. Delay in diagnosis of spinal muscular atrophy: A systematic literature review. Pediatr Neurol 2015;53(4):293-300. https://doi.org/10.1016/j.pediatrneurol.2015.06.002 [ Links ]
3. Labrum R, Rodda J, Krause A. The molecular basis of spinal muscular atrophy (SMA) in South African black patients. Neuromuscul Disord 2007;17(9-10):684-692. https://doi.org/10.1016/j.nmd.2007.05.005 [ Links ]
4. Verhaart IEC, Robertson A, Wilson IJ, et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis 2017;2(1):124. https://doi.org/10.1186/s13023-017-0671-8 [ Links ]
5. DAmico A, Mercuri E, Tiziano FD, Bertini E. Spinal muscular atrophy. Orphanet J Rare Dis 2011;6:71. https://doi.org/10.1186/1750-1172-6-71 [ Links ]
6. Chen H. Atlas of genetic diagnosis and counseling. Cham: Springer Nature Switzerland AG, 2006. https://doi.org/10.1007/978-1-4614-1037-9 [ Links ]
7. Kolb SJ, Kissel JT. Spinal Muscular Atrophy. Neurol Clin 2015;33(4):831-846. https://doi.org/10.1016/j.ncl.2015.07.004 [ Links ]
8. Al Dakhoul S. Very severe spinal muscular atrophy (Type 0). Avicenna J Med 2017;(1):32-33. [ Links ]
9. Hamilton G, Gillingwater TH. Spinal muscular atrophy: Going beyond the motor neuron. Trends Mol Med 2013;19(1):40-50. https://doi.org/10.1016/j.molmed.2012.11.002 [ Links ]
10. Wilmshurst JM, Reynolds L, Van Toorn R, et al. Spinal muscular atrophy in black South Africans: Concordance with the universal SMN1 genotype. Clin Genet 2002;62(2):165-168. [ Links ]
11. Stevens G, Yawitch T, Rodda J, Verhaart S, Krause A. Different molecular basis for spinal muscular atrophy in South African black patients. Am J Med Genet 1999;86(5):420-426. [ Links ]
12. Arnold DW, Kassar D, Kiseel JT. Spinal muscular atrophy: Diagnosis and management in a new therapeutic era. Muscle Nerve 2015;51(2):157-167. https://doi.org/10.1002/mus.24497 [ Links ]
13. Desguerre I. Spinal muscular atrophy. Eur J Paediatr Neurol 2017;21:e13. [ Links ]
14. Quansah E, Karikari TK. Motor neuron diseases in sub-Saharan Africa: The need for more population-based studies. Biomed Res Int 2015;2015:298409. https://doi.org/10.1155/2015/298409 [ Links ]
15. Moosa A, Dawood A. Spinal muscular atrophy in African children. Neuropediatrics 1990;21(1):27-31. https://doi.org/10.1055/s-2008-1071453 [ Links ]
16. Vorster E, Essop FB, Rodda JL, et al. Spinal muscular atrophy in the black South African population: A matter of rearrangement? Front Genet 2020;11:54. https://doi.org/10.3389/fgene.2020.00054 [ Links ]
 Correspondence:
Correspondence:
K Flack
katherine.flack@gmail.com
Accepted 17 January 2022


{kind=link}
{kind=link}
{kind=link}