










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Child Health
On-line version ISSN 1999-7671
Print version ISSN 1994-3032
S. Afr. j. child health vol.16 n.4 Pretoria Dec. 2022
http://dx.doi.org/10.7196/SAJCH.2022.v16i4.1931
CASE REPORT
Two South African patients with PGAP3-related Mabry syndrome with unusually low alkaline phosphatase levels
S Moosa
MMed, PhD; Division of Molecular Biology and Human Genetics, Faculty of Medicine and Health Sciences, Biomedical Sciences, Stellenbosch University and Medical Genetics, Tygerberg Hospital, Cape Town, South Africa
ABSTRACT
Hyperphosphatasia with mental retardation syndrome (HPMRS), also known as Mabry syndrome, is an autosomal recessive disease that is associated with inherited glycosylphosphatidylinositol (GPI) deficiencies. This genetically heterogeneous disorder can be caused by variants in seven genes that encode molecules of the glycosylphosphatidylinositol (GPI)-anchor biosynthesis pathway, namely PIGL, PIGO, PIGV, PIGW, PIGY, PGAP2 and PGAP3. Recently, a pathogenic variant in PGAP3 was identified in 3 unrelated South African patients with HMPRS. Here, two further patients with the exact variant in PGAP3 are described. Classically, HMPRS is associated with elevated alkaline phosphatase (ALP) levels. Interestingly, these two patients had unusually low ALP levels at initial presentation. This is an important observation, as the ALP level is often used as a screening test to decide whether to proceed to confirmatory genetic testing. These patients illustrate that in PGAP3-related Mabry syndrome, ALP levels can be low, albeit a rare finding. Hence, a high suspicion for the disorder should be maintained in patients with typical facial dysmorphic features and severe neurodevelopmental delay, even in the absence of elevated ALP.
Hyperphosphatasia with mental retardation syndrome (HPMRS) (OMIM239300), also known as Mabry syndrome, is an autosomal recessive disorder which was first described in 1970 in 3 patients who presented with developmental delays and intellectual disability, seizures and elevated serum alkaline phosphatase (ALP).[1] Patients with HMPRS typically present with severe intellectual disability, characteristic facial dysmorphic features, and elevated ALP levels. Other features are variable and include seizures, cleft palate, brachytelephalangy, anorectal and gastrointestinal anomalies, as well as cardiac malformations.[3]
HMPRS is genetically heterogeneous, with seven genes involved in GPI-anchor synthesis currently known to cause the disorder, namely: PIGL (OMIM605947), PIGV (OMIM610274), PIGW (OMIM610275), PIGY (OMIM610662), PIGO (OMIM614730), PGAP2 (OMIM614207) and PGAP3 (OMIM615716) have been described in patients with HPMRS.
Recently, 3 unrelated South African (SA) patients with typical features of HMPRS were described to have a homozygous pathogenic variant in PGAP3 (NM_0033419.5: c.557G>T; p.Arg186Thr); thus confirming a diagnosis of HMPRS type 4 in them.[1] All 3 had massively elevated ALP levels. Here, 2 further SA patients with the same pathogenic variant in PGAP3 are described. They share the typical clinical features with other patients previously described. However, they had abnormally low levels of ALP at presentation.
Both patients were enrolled in our Undiagnosed Disease Programme at Stellenbosch University (HREC ref. no. N18/03/031). Informed consent was obtained from the parents for whole-exome sequencing, and for publication of results with corresponding clinical information and photographs. ALP was measured using standard NHLS laboratory protocols.
Case reports
Patient 1 is the only child to his non-consanguineous parents. His older maternal half-sister is well. There are no known conditions in the family. He presented with facial dysmorphic features (Figs 1A and 1B), cleft palate and developmental delay. At the age of 18 months, he was not sitting independently and was not saying any words. He also had bilateral undescended testes. Previous tests including a karyotype and a chromosomal microarray were normal. He was evaluated at our Medical Genetics clinic and the clinical diagnosis of HMPRS was made. His initial ALP at that time was 12 U/L (normal range 104 - 345 U/L). In light of the strong clinical suspicion, we performed whole-exome sequencing on DNA from the patient and confirmed that he was homozygous for the PGAP3 c.557G>T (p.Arg186Thr) variant. When he came back for results delivery at 2 years old, his ALP level was high (862 U/L).
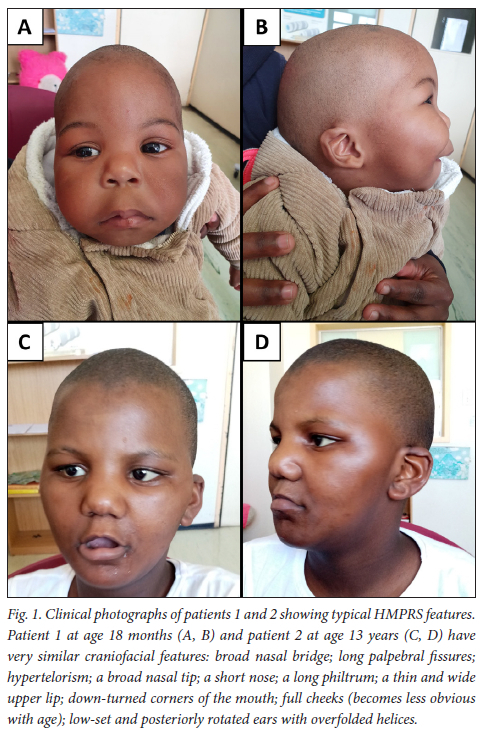
Patient 2 was the only child of non-consanguineous parents. She had two younger maternal half-siblings who were well. The rest of the family history was noncontributory. She presented to our Medical Genetics clinic for an evaluation of her developmental delay and dysmorphic features (Figs 1C and 1D). She was born with a cleft palate, which was repaired. All her milestones were delayed. At 13 years old, she was not yet toilet trained, could only say a few single words, was aggressive and hyperactive. She attended a special school but was making little progress. Her facial features were those of classic HMPRS. Duo whole-exome sequencing was performed on her and her mother and confirmed the diagnosis. The patient was homozygous for the PGAP3 c.557G>T (p.Arg186Thr) variant, while the mother was heterozygous. Her ALP level was 16 U/L (normal range 50 - 162 U/L).
Discussion
Both patients had classic dysmorphic features associated with HMPRS, along with the developmental delay and other well-known malformations in this syndrome. However, their baseline ALP levels were abnormally low, which is a very rare finding in HMPRS. Previous studies have shown that a minority of patients with PGAP3-related HMPRS can have low ALP levels.™ However, until now, we have used an elevated ALP level as a screening test, before progressing to genetic testing. Indeed, all previously described SA patients with HMPRS had elevated ALP levels.[1]
This observation of low ALP is important, as Sanger sequencing for the PGAP3 c.557G>T (p.Arg186Thr) variant is now available as a diagnostic test through the NHLS at the University of Cape Town (UCT). We therefore urge clinicians to have a high suspicion for HMPRS in patients with the typical facial gestalt, even if ALP levels are initially low. Confirming a diagnosis of this recessively inherited syndrome allows for accurate genetic counselling, prognostication, and even prenatal testing if desired.
Declaration. None.
Acknowledgements. Thanks are extended to the families for their enthusiastic participation in the Undiagnosed Disease Programme. We thank the Centre for High Performance Computing (CHPC; http://www.chpc.ac.za) for computational resources. We also thank all the clinicians involved in the care of these patients, and the molecular genetics laboratory team at NHLS (UCT) for developing a diagnostic test for HMPRS4.
Author contributions. Sole author.
Funding. Whole-exome sequencing was performed at 3billion (Seoul, Republic of Korea). The work reported herein was also made possible through funding by the South African Medical Research Council (SAMRC) through its Division of Research Capacity Development under the Early Investigators Programme from funding received from the South African National Treasury. The content hereof is the sole responsibility of the authors and does not necessarily represent the official views of the SAMRC.
Conflicts of interest. None.
References
1. Bezuidenhout H, Bayley S, Smit L, et al. Hyperphosphatasia with mental retardation syndrome type 4 in three unrelated South African patients. Amer J Med Genet Part A 2020;182(10):2230-2235. https://doi.org/10.1002/ajmg.a.61797 [ Links ]
2. Mabry CC, Bautista A, Kirk RFH, Dubilier LD, Braunstein H, Koepke JA. Familial hyperphosphatasia with mental retardation, seizures, and neurologic deficits. J Pediatr 1970;77:74-85. [ Links ]
3. Knaus A, Pantel JT, Pendziwiat M, et al. Characterisation of glycosylphosphatidylinositol biosynthesis defects by clinical features, flow cytometry, and automated image analysis. Genome Med 2018;10(1):3. https://doi.org/10.1186/s13073-017-0510-5 [ Links ]
 Correspondence:
Correspondence:
S Moosa
shahidamoosa@sun.ac.za
Accepted 28 October 2021

