










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Child Health
versão On-line ISSN 1999-7671
versão impressa ISSN 1994-3032
S. Afr. j. child health vol.16 no.2 Pretoria Jun. 2022
http://dx.doi.org/10.7196/sajch.2022.v16i2.1872
RESEARCH
Primary hyperoxaluria: The Baragwanath experience
C L ChangI, II; K L PetersenIII, IV, V; A M CilliersVI, VII, VIII; U K KalaIX, X, XI
IFCPaed (SA); Department of Paediatrics, Chris Hani Baragwanath Academic Hospital, and the Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIFCPaed (SA); Department of Paediatrics, Faculty of Health Sciences, University of Witwatersrand, Johannesburg, South Africa
IIICert Nephrology (SA) (Paed); Department of Paediatrics, Chris Hani Baragwanath Academic Hospital, and the Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IVCert Nephrology (SA) (Paed); Department of Paediatric Nephrology, Chris Hani Baragwanath Academic Hospital, and the Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
VCert Nephrology (SA) (Paed); Department of Paediatrics, Faculty of Health Sciences, University of Witwatersrand, Johannesburg, South Africa
VIFCPaed; Department of Paediatrics, Chris Hani Baragwanath Academic Hospital, and the Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
VIIFCPaed; Department of Paediatric Cardiology, Chris Hani Baragwanath Academic Hospital, and the Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
VIIIFCPaed; Department of Paediatrics, Faculty of Health Sciences, University of Witwatersrand, Johannesburg, South Africa
IXCert Nephrology (SA) (Paed); Department of Paediatrics, Chris Hani Baragwanath Academic Hospital, and the Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
XCert Nephrology (SA) (Paed); Department of Paediatric Nephrology, Chris Hani Baragwanath Academic Hospital, and the Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
XICert Nephrology (SA) (Paed); Department of Paediatrics, Faculty of Health Sciences, University of Witwatersrand, Johannesburg, South Africa
ABSTRACT
BACKGROUND: Primary hyperoxaluria (PH) is a rare autosomal recessive condition characterised by defects in the metabolism of glyoxylate which leads to excess oxalate production. It is an important disease to diagnose as it can progress to kidney failure (KF
OBJECTIVE: To describe the characteristics, diagnosis and management of PH in South Africa and to identify any determinants of KF and death
METHOD: A retrospective study of all children younger than 16 years of age, diagnosed with PH at the Paediatric Renal Unit, Chris Hani Baragwanath Academic Hospital, from 1984 - 2017
RESULTS: A total of 24 patients were identified, of which 20 records were available for complete analysis. The median age of presentation was 6.0 years. The common clinical presentations were urolithiasis (90%), KF (85%), nephrocalcinosis (75%), urinary tract infections (55%) and haematuria (30%). Nephrocalcinosis was better detected on abdominal radiograph compared with ultrasonography. Both nephrocalcinosis (p=0.009) and haematuria (p=0.018) were significantly associated with KF. Five patients had A112D genetic mutation in the AGXT. Fourteen received dialysis and four were transplanted. The mortality rate in this study was 58.3%
CONCLUSION: Clinicians should have a high index of suspicion for PH in patients presenting with haematuria, urolithiasis and KF. This study supports the measurement of urine oxalate levels and abdominal radiographs in screening for PH in children presenting in KF
Primary hyperoxaluria (PH) is a rare autosomal recessive condition characterised by defects in the metabolism of glyoxylate which leads to an increased oxalate production and deposition.[1] The most common and severe form is type 1 PH (PH1), due to a genetic mutation in the enzyme, alanine-glyoxylate aminotransferase (AGT) found in the liver peroxisomes.[1] PH1 rapidly progresses to kidney failure (KF), which includes the more severe presentation of infantile PH subtype.[2] The exact prevalence and incidence of PH is unknown.[1]
The age of presentation can range from birth to the sixth decade of life.[1] The most common presentations are urolithiasis, with or without nephrocalcinosis, urinary tract infections (UTIs), haematuria and KF.[3-6] Systemic oxalosis is a result of the deposition of insoluble calcium oxalate crystals in any organ, which usually develops once the glomerular filtration rate (GFR) falls below 40 mL/ min/1.73 m2.[7] The diagnostic work-up includes a 24-hour urine or spot urine analysis for oxalate crystals, genetic mutation analysis, imaging and histology.[1,7] Management of PH includes high fluid intake of 2 - 3 L/m2 of body surface area per day, alkalinisation of the urine and pyridoxine supplementation.[1,7] Combined peritoneal (PD) and haemodialysis (HD) is also required for the management of PH, but the definitive treatment for PH1 is a combined liver and kidney transplant.[1]
Although primary hyperoxaluria is a rare genetic disorder, the clinical data on PH has been well documented in developed countries.[3,8,9] There are data emerging from developing countries with cohorts of 18 patients from Oman, [10] 26 from Egypt,[6] 44 from Tunisia,[5] and 70 from Jordan.[4] However, no studies have yet been published in sub-Saharan Africa, including South Africa (SA). This study aimed to describe the presentation, diagnosis and treatment of PH at Chris Hani Baragwanath Academic Hospital (CHBAH) in SA, and to identify any determinants of KF and death.
Method
The study population was selected from the Paediatric Renal Unit at Chris Hani Baragwanath Academic Hospital (CHBAH), which is a tertiary academic hospital in Gauteng affiliated to the University of the Witwatersrand. This unit serves the population of Soweto but also receives patients referred from other provinces, as well as, neighbouring countries of SA.
This was a retrospective descriptive study spanning 33 years, 1 January 1984 - 31 December 2017, of all patients younger than 16 years of age and diagnosed with PH. The diagnosis of PH was made if any one of the following was fulfilled: high urine oxalate levels in 24-hour urine collection (>0.7 mmol/1.73 m2)[1] or spot urine analysis in keeping with hyperoxaluria[1] or birefringent oxalate crystals on histology or genetic confirmation.
Demographics, diagnostic tests, management and outcome data were obtained from the records and further analysed for any association with KF and death. For the definition of the different variables, please refer to the supplementary file (url:XX).
The categorical data were presented as frequencies and percentages and continuous data as medians with inter-quartile ranges (IQRs). A Pearson's chi-squared test or Fisher's exact test was used to analyse categorical variables for association with KF and death. For continuous variables that was not normally distributed, a Mann-Whitney U test was used. The data were analysed at the 95% confidence interval (CI) and p-value <0.05 was considered significant. The software used was STATISTICA version 13.3 (TIBCO Software Inc., USA). Ethics approval was obtained from the Human Research Ethics Committee of the University of the Witwatersrand (ref. no. M170423) as well as the medical advisory committee at CHBAH. Approval was also granted by the head of the renal unit to utilise the records.
Results
A total of 24 patients were identified with PH, of which 20 records were available for complete analysis. The data available for the four incomplete records were included in the analysis.
Demographics
The median (IQR) age of presentation was 72.0 (44.0 - 108.0) months (Table 1). Nineteen patients (95.0%) were South African, and of African descent, while one was from Swaziland, of British and caucasian descent. Of the 11 patients from Gauteng, 81.8% were from outside Soweto. There was no history of consanguinity or previous family history of kidney disease.

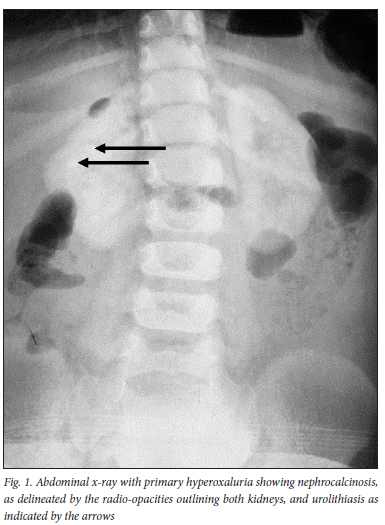
As seen in Table 2, the most common clinical presentation was urolithiasis (90.0%) and KF (85.0%). The median (IQR) estimated GFR (eGFRcr) was 6.1 (4.2 - 9.0) ml/min/ 1.73 m2. One patient had stage four (CKD) and two patients had normal kidney functions. Figure 1 depicts nephrocalcinosis and urolithiasis detected by plain abdominal radiograph. Most of the patients had more than one test performed for the diagnosis of PH. The median (IQR) 24-hour urine oxalate excretion was 1.4 (0.8 - 2.0 mmol/1.7 m2/day and 323.6 (232.5 - 594.1) μmol/mmol for spot urine oxalate to creatinine ratio. More spot urine samples were collected compared with 24-hour urine oxalate samples. However, the positivity rate was higher with 24-hour urine collections (87.5%) compared with the spot urine samples (76.9%). Bone marrow aspirate and trephine (BMAT) was performed on 16 patients, of which five (31.3%) had confirmed oxalate crystals under polarised light. Kidney biopsy was done on 11 of the patients, including one postmortem biopsy, and all confirmed oxalate crystals under polarised light.

Echocardiography was done on 15 patients, eight (53.3%) of which had normal findings and seven (46.7%) had abnormal findings: two patients (13.3%) had left ventricular hypertrophy, two patients (13.3%) had small pericardial effusions (range 4 - 8 mm), one patient (6.7%) had diastolic dysfunction, and one patient (6.7%) had a patent ductus arteriosus (PDA). Lastly, echo-bright myocardium was observed in one patient (6.7%), but no speckling was observed. There were seven electrocardiograms (ECG) available for analysis, four were normal but three patients had prolonged, corrected QT intervals. Of the three patients, two had documented hypocalcaemia.
Genetic studies were done on six patients, and all had confirmed type 1 PH. Five of the patients were homozygous for A112D mutation and the other had a heterozygous mutation for two different alleles, c335C>A (p.A112D) and c.473C>T (p.S158L).
On initial presentation, 15 (75.0%) patients had nephrocalcinosis on abdominal radiograph. Of the remaining five, one had urolithiasis without nephrocalcinosis; two were not available and two of the patients, who had normal kidney function, showed normal plain abdominal radiographs. This contrasts with the 17 ultrasounds that were performed, where only five (29.4%) ultrasounds confirmed the presence of medullary or cortical nephrocalcinosis. Of the remaining 12 ultrasounds, five reported hyperechoic kidneys and the remaining seven sonars did not identify nephrocalcinosis. Nephrocalcinosis was significantly associated with KF (p=0.009).
Management
Routine management included alkalinisation of urine, high fluid intake and pyridoxine. The median (IQR) dose of pyridoxine was 6(5- 10) mg/kg/dose. Response to pyridoxine treatment was not assessed in this study. A gastrostomy tube was inserted for a 7-month-old infant to ensure adequate fluid and protein intake. Three patients had endoscopic lithotripsy by the urologist prior to referral to the renal unit.
A total of 14 patients received dialysis and four patients died prior to the initiation of dialysis. Of the 11 patients that were referred for transplantation, four had been transplanted, of which two received combined liver-kidney transplantation and the other two had kidney transplantation. Of the remaining seven patients, two were waiting to be listed, four patients demised prior to transplantation, and one was lost to follow-up. In this study, 14 of the 24 patients demised resulting in a mortality rate of 58.3%.
There were significant associations between the following factors with KF: nephrocalcinosis (p=0.009), and haematuria (p=0.018). However, no significant determinants of death were identified.
Discussion
This is one of the first studies reporting on PH in SA. It highlighted the important differences in the presentation of patients in this cohort compared with other countries, such as North Africa and the Middle East. The most notable difference was the age of presentation. There was no history of consanguinity in this study, compared with the North African[5] and Middle Eastern cohorts.[4,6,10] The high prevalence of PH seen in the North African and Middle Eastern countries may be due to the high rates of consanguinity.[1]
In this study, the median age of onset of first symptoms was 67.5 months (5.6 years), which is comparable with the Dutch cohort (median age of 6.0 years).[9] The median age of presentation was 72 months (6 years) which is much older compared to the Egyptian cohort of 3 years of age.[6] The difference in age of presentation was a result of the higher proportion of infantile PH (35.3%) in the Egyptian cohort[6] compared with this study cohort of 4.2%.
The most common clinical presentation in this study was urolithiasis, KF and nephrocalcinosis, which was similar to the other international cohorts.[3,6,9,10] In this cohort, nephrocalcinosis was identified more often on plain abdominal radiographs compared with ultrasound, and as reported in other studies.[11,12]
It has been shown in a few studies that cortical nephrocalcinosis is associated with rapid progression to KF.[5,6,8,9,13] In this cohort, the presence of nephrocalcinosis was associated with KF (p=0.009). The poor recognition of nephrocalcinosis on sonography may be due to the low diagnostic yield and the high inter-observer variability in this imaging modality.[14] In addition, the hyperechoic renal parenchyma on ultrasound could represent nephrocalcinosis, but not interpreted as such. This highlights the importance of plain abdominal radiographs, as well as the utilisation of more than one imaging modalities to improve nephrocalcinosis-diagnosis in PH.
The percentage of positive results were higher in the 24-hour urine collections (87.5%) compared with the spot urine samples (76.9%). A 24-hour urine collection is the gold standard diagnostic test and is preferred over a spot urine sample, as it is more sensitive and specific.[15] However, a 24-hour urine collection is technically more challenging, and requires accurate timing and collection of all urine passed during that time period.[16] A spot urine oxalate creatinine is also more practical in younger children.[15]
The decline in GFR typically results in reduction in urine oxalate excretion.[16] However, this was not evident in this study. Urinary oxalate levels were diagnostic in 11 patients with eGFRcr < 30 mL/ min/1.73 m2, and remains an important diagnostic tool for PH even in advanced stages of CKD. Haematuria was shown to be significantly associated with KF (p=0.018), showing the importance of routine urine dipsticks.
Genetic testing was performed on six of the patients, of which five had homozygous A112D mutation (type 1 PH). This mutation was previously identified in two unrelated patients, one from Botswana and one South African.[17] A112D is most likely a common mutation in the Southern African population as previously suggested.[17] One patient was heterozygous for two different alleles, c335C>A (p.A112D) and c.473C>T (p.S158L).
Given the small sample size there was no significant association noted between the genetic mutations with KF (p=1.0). Interestingly, of all the genetically-confirmed PH1 patients, four had KF while one had normal kidney function. The patient with the normal kidney function and type 1 PH likely had a slow progression of the disease, supporting the notion that simply having the genotype does not necessarily translate into full phenotypic expression. Further studies would need to be done to identify phenotypic correlation to homozygous A112D mutation. It was not possible to determine the PH subtypes of the remaining 11 patients, who had presented with KF, without genetic analysis.
Histology remains an important tool in the diagnosis of PH, particularly in anuric patients where measuring urine oxalate levels is not possible. Kidney biopsy appeared to be more relaible than BMAT in this cohort. There was no significant association between systemic oxalosis and KF, or death.
The cardiac abnormalities noted in the study population, including left ventricular hypertrophy and diastolic dysfunction, could be related to the hypertension seen in chronic kidney disease, but it could also be attributed to oxalate deposition as observed in another study.[18] Increased cardiac echogenicity has also been described in patients with PH as a result of deposition of oxalate crystals,[19] which was observed in one study patient.
There was one study patient that had a prolonged QT interval with normal calcium levels. This could be a result of oxalate deposition as arrhythmias have been described in PH patients.[18] There was no significant association between the cardiac manifestations and KF, or death. Non-invasive screening for cardiac involvement in all patients with PH is recommended. Larger studies would be valuable in characterizing the cardiac manifestations of PH.
A large proportion of the study population were referred from the North West (36.8%) and of those that were from Gauteng, 81.8% were from outside of Soweto, the drainage area for CHBAH. Transportation costs incurred by patients may account for as much as 27.1% of the average family income.[20] In addition to the transportation costs, the need for families to relocate to access dialysis and further treatment also results in high social strain and disruption to the family unit, as well as, daily life.
It is possible that the mortality rate in this study was higher than 58.3%, as three of the four patients that defaulted follow-up had eGFRcr <15 mL/min/1.73 m2. This mortality was higher compared to the Egyptian cohort of 42.3%, of which 65.4% of the patients had KF.[6] The higher mortality rate in this study was likely due to late presentation, as well as, the higher portion of KF (85.0%) in this cohort.
This retrospective study was limited by the small sample size, the lack of availability and incompleteness of the records in four children. In this cohort, the lack of significance between systemic oxalosis with KF or death and the determinants of death, was likely due to the small sample size. Logistic models for analysis of the determinants of KF and death could not be done due to the small sample size. There is a referral bias given that only patients that were referred to the renal unit were included. The milder and more severe types of PH may be missed, as the former may be asymptomatic, and the latter die before referral or presentation. Despite these limitations, this study is the first in SA to document PH in children, and future studies may add to the genetic data.
Conclusion
Clinicians should have a high index of suspicion for PH in patients presenting with haematuria, urolithiasis and KF. This study supports the measurement of urine oxalate levels and abdominal radiographs in screening for PH in children presenting in KF.
Declaration. This research report was completed in fullfillment of MMed (Paed) at the University of the Witwatersrand.
Acknowledgements. The authors wish to acknowledge the clerks, the data capturers who made data collection possible, the medical and nursing staff invloved in the care the patients, and statisticians at Philip Tobias, University of Witwatersrand.
Author contributions. CLC: conception of research question, data collection, data analysis, manuscript preparation and revision; KLP and UKK: conception of research question, data analysis, revision and approval of final manuscript. AMC: data collection and analysis and approval of final manuscript.
Funding. None.
Conflicts of interest. None.
References
1. Cochat P, Rumsby G. Primary hyperoxaluria. N Engl J Med 2013;369(7):649-658. https://doi.org/10.1056%2Fnejmra1301564 [ Links ]
2. Cochat P, Liutkus A, Fargue S, Basmaison O, Ranchin B, Rolland MO. Primary hyperoxaluria type 1: Still challenging! Pediat Nephrol 2006;21(8):1075-1081. https://doi.org/10.1007%2Fs00467-006-0124-4 [ Links ]
3. Hoppe B, Latta K, von Schnakenburg C, Kemper MJ. Primary hyperoxaluria-the German experience. Am J Nephrol 2005;25(3):276-281. https://doi.org/10.1159%2F000086358 [ Links ]
4. Almardini RI, Alfarah MG, Salaita GM. The clinical pattern of primary hyperoxaluria in pediatric patient at Queen Rania Abdulla Children Hospital. Arab J Nephrol Transplant 2014;7(2):119-123. [ Links ]
5. Gargah T, Khelil N, Youssef G, Karoui W, Lakhoua MR, Abdelmoula J. Primary hyperoxaluria type 1 in Tunisian children. Saudi J Kidney Dis Transpl 2012;23(2):385-390. https://pubmed.ncbi.nlm.nih.gov/22382246/ [ Links ]
6. Soliman NA, Nabhan MM, Abdelrahman SM, et al. Clinical spectrum of primary hyperoxaluria type 1: Experience of a tertiary center. Nephrol Ther 2017;13(3):176-182. https://doi.org/10.1016%2Fj.nephro.2016.08.002 [ Links ]
7. Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int 2009;(75): 1264-1271. https://doi.org/10.1038%2Fki.2009.32 [ Links ]
8. Takayama T, Nagata M, Ichiyama A, Ozono S. Primary hyperoxaluria type 1 in Japan. Am J Nephrol 2005;25(3):297-302. https://doi.org/10.1159%2F000086361 [ Links ]
9. van Woerden CS, Groothoff JW, Wanders RJ, Davin JC, Wijburg FA. Primary hyperoxaluria type 1 in The Netherlands: prevalence and outcome. Nephrol Dial Transplant 2003;18(2):273-279. https://doi.org/10.1093%2Fndt%2F18.2.273 [ Links ]
10. Al Riyami MS, Al Ghaithi B, Al Hashmi N, Al Kalbani N. Primary hyperoxaluria type 1 in 18 children: genotyping and outcome. Int J Nephrol. 2015:1-6. https://doi.org/10.1155%2F2015%2F634175 [ Links ]
11. Singh DR, Sagade SN, Kamat MH, Deshpande RB, Shah BV. Oxalosis with nephrocalcinosis. Nephrol Dial Transplant 2000;15(1):124-125. https://doi.org/10.1093%2Fndt%2F15.1.124 [ Links ]
12. Orazi C, Picca S, Schingo PMS, Fassari FM, Canepa G. Oxalosis in primary hyperoxaluria in infancy: Report of a case in a 3-month-old baby. Follow-up for 3 years and review of literature. Skeletal Radiol 2009;38(4):387-391. https://doi.org/10.1007%2Fs00256-008-0625-2 [ Links ]
13. Tang XJ, Bergstralh EJ, Mehta RA, Vrtiska TJ, Milliner DS, Lieske JC. Nephrocalcinosis is a risk factor for kidney failure in primary hyperoxaluria. Kidney Int 2015;87(3):623-631. https://doi.org/10.1038%2Fki.2014.298 [ Links ]
14. Cheidde L, Ajzen SA, Langen CHT, Christophalo D, Heilberg IP. A critical appraisal of the radiological evaluation of nephrocalcinosis. Nephron Clin Pract 2007;106(3):119-124. https://doi.org/10.1159%2F000102999 [ Links ]
15. Hashmi SB, Jafri L, Majid H, Talati J, Aziz W, Khan AH. Relationship of spot urine oxalate to creatinine ratio and 24 hours urinary oxalate excretion in patients with urolithiasis. Ann Med Surg (Lond) 2020;60:330-333. https://doi.org/10.1016/j.amsu.2020.11.002 [ Links ]
16. Clifford-Mobley O, Tims C, Rumsby G. The comparability of oxalate excretion and oxalate: creatinine ratio in the investigation of primary hyperoxaluria: review of data from a referral centre. Ann Clin Biochem 2015;52(1):113-121. https://doi.org/10.1177%2F0004563214529937 [ Links ]
17. Coulter-Mackie MB, Tung A, Henderson HE, Toone JR, Applegarth DA. The AGT gene in Africa: a distinctive minor allele haplotype, a polymorphism (V326I), and a novel PH1 mutation (A112D) in Black Africans. Mol Genet Metab. 2003;78(1):44-50. https://doi.org/10.1016%2Fs1096-7192%2802%2900204-4 [ Links ]
18. Mookadam F, Smith T, Jiamsripong P, et al. Cardiac Abnormalities in Primary Hyperoxaluria. Circ J 2010;74(11):2403-2409. https://doi.org/10.1253%2Fcircj.cj-10-0107 [ Links ]
19. Yoshioka J, Park YD, Tanaka Y, et al. Echocardiographic Features in a Patient with Primary Oxalosis. Echocardiography 2001;18(7):599-602. https://doi.org/10.1046%2Fj.1540-8175.2001.00599.x [ Links ]
20. Bello A, Sangweni B, Mudi A, Khumalo T, Moonsamy G, Levy C. The financial cost incurred by families of children on long-term dialysis. Perit Dial Int 2018;38(1):14-17. https://doi.org/10.3747%2Fpdi.2017.00092 [ Links ]
 Correspondence:
Correspondence:
C-L Chang
chihluochang@gmail.com
Accepted 7 July 2021

