










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Child Health
versão On-line ISSN 1999-7671
versão impressa ISSN 1994-3032
S. Afr. j. child health vol.15 no.3 Pretoria Set. 2021
http://dx.doi.org/10.7196/SAJCH.2021.v15.i3.1820
CASE REPORT
Alagille syndrome and hereditary von Willebrand disease: A rare co-occurrence
M J DempsterI; B F JacobsonII; P WalabhIII
IMB BCh; Vaccines and Infectious Diseases Analytical Research Unit, Johannesburg, South Africa
IIPhD; Department of Clinical Haematology, Charlotte Maxeke Johannesburg Academic Hospital and Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIIFC Paed (SA), Cert Gastroenterol (SA); Department of Paediatrics and Child Health, Charlotte Maxeke Johannesburg Academic Hospital and Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
Alagille syndrome (ALGS) and von Willebrand disease (vWD) are both genetic conditions. An individual presenting with two independent genetic conditions is rare and there are no reported cases of these two conditions occurring in a single individual. ALGS often manifests with cholestasis and could lead to end-stage liver disease and associated complications, such as a variceal bleed. vWD is a bleeding disorder. This case report describes the rarity of these two genetic conditions and the management of a potential life-threatening bleed secondary to oesophageal varices and high bleeding risk.
Case presentation
A 17-year-old female patient of Indian ethnicity diagnosed with Alagille syndrome (ALGS) complicated by pulmonary stenosis and von Willebrand disease (vWD) presented with hypochromic anaemia and, upon further examination, splenomegaly. The underlying pathologies, combined with the observed splenomegaly and hypochromic anaemia, raised concern of oesophageal varices and a potential life-threatening haemorrhage.
The patient had been diagnosed with ALGS as an infant based on phenotypic criteria that included cholestasis, facial features, a butterfly vertebra at T11 and peripheral pulmonary artery stenosis. The most recent liver function results showed normal bilirubin levels and mild derangement of transaminases. Ultrasound examination reported the spleen measuring 12.7 cm, but there was no sonographic evidence of portal hypertension (Fig. 1).
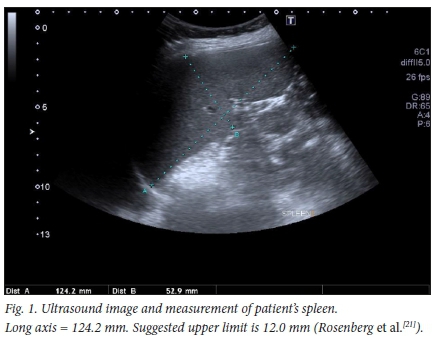
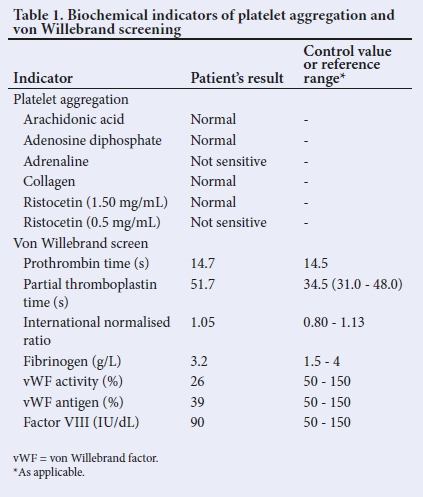
The patient's diagnosis of congenital vWD was based on a first-degree relative (mother) and confirmed through blood tests measuring von Willebrand factor (vWF) activity, vWF antigen level and factor VIII (FVIII) activity (Table 1). The diagnosis indicated a severe form of the disease. Clinically, the patient had not had a life-threatening bleeding episode. The patient's vWD symptoms had been managed with desmopressin (DDAVP), with excellent response (Table 2).

An oesophagogastroduodenoscopy was performed under conscious sedation to exclude oesophageal varices. No DDAVP cover was given prior to the procedure. The examination showed no visible varices, although a small hiatus hernia was noted.
It was concluded that the hypochromic anaemia was secondary to heavy menstrual bleeding. The duration of the patient's menstruation was ~7 days and associated with heavy bleeding. She had previously been treated with DDAVP during her menstruation. Oral iron supplementation was added (as her serum iron levels were towards the lower limit of normal and serum saturation of iron was low). After 4 months of iron supplementation, the patient's anaemia had resolved.
Discussion
Von Willebrand disease
vWD is a bleeding disorder that affects platelet adhesion and aggregation. It is generally an inherited disorder, with ~1 in 10 000 people experiencing symptomatic disease.[1] An acquired form can occur, associated with certain medical conditions (such as lymphoproliferative disorders, autoimmune states, endocrine disorders and various others).
The affected factor in the disease is vWF, which is synthesised in the endothelium and megakaryocytes, and stored as Weibel-Palade bodies in the endothelium and as alpha granules in platelets. vWF from the endothelium is released into the subendothelium and then into the plasma in response to physiological stressors. Alpha granules also release vWF in response to various agonists.[2] It binds to collagen at sites of vascular injury, mediates platelet adhesion and aggregation, and serves as a carrier protein for coagulation factor VIII.[3] vWD therefore occurs when there is a deficiency in the quality or quantity of vWF.
There are three forms of vWD, namely: hereditary, acquired, and the pseudo- or platelet type. The vWF gene is found on the short arm of chromosome 12 (12p13.2). The hereditary types are classified as type 1, 2 or 3, with various subtypes; the platelet type, which is also inherited, is considered a separate entity. Types 1 and 2 are inherited as an autosomal dominant trait, whereas type 3 is autosomal recessive. Types 1 and 3 affect vWF quantitatively, whereas type 2 presents as a functional defect.
Diagnosis relies on a combination of personal or family history and laboratory evidence. Various stressors, such as pregnancy, hormonal fluctuations, hormonal therapies, infection, surgery and strenuous exercise, affect vWF levels. These stressors alter plasma levels of oestrogen, vasopressin, growth hormone and adrenergic stimuli, which, in turn, affect vWF levels.
Repeat testing at intervals of two or more weeks is advised to confirm diagnosis of vWD.[4] Screening for vWD includes measuring activated partial thromboplastin time, determining the platelet count and running the platelet function analyser test (PFA 100), which measures both platelet adhesion to collagen and platelet aggregation. For accurate diagnosis of vWD, the level of vWF present in the plasma, its efficacy in binding to platelets in the presence of the antibiotic ristocetin and the level of factor VIII need to be determined. Factor VIII levels assist with classifying patients according to the mentioned types and subtypes.[1] vWF multimer analysis, which offers a qualitative visual assessment of the size spectrum and banding pattern of vWF multimers in the plasma,[5] can be used to identify variants of type 2 vWD.
Treatment and prophylactic management depend on the severity of the disease and whether invasive procedures or surgery are being performed. Mild vWD is often managed with DDAVP, with response determined by laboratory testing. If DDAVP treatment is inadequate, replacement therapy with purified vWF-containing concentrates is indicated. Secondary long-term prophylaxis with concentrates is used to manage severe forms of vWD.[6]
A rare complication of vWD is angiodysplasia, which can result in severe intractable gastrointestinal bleeding. Angiodysplasia is most commonly found in the caecum and ascending colon, although cases of small-intestine and stomach involvement have been reported.
Alagille syndrome
ALGS is an autosomal dominant genetic disorder that affects multiple systems - mainly the liver, heart, skeleton, face and eyes. Although inheritance is autosomal dominant, the syndrome is associated with variable expression, reduced penetrance and possible germline mosaicism. The prevalence of ALGS is reported to be ~1 : 30 000.[7]
The inherited pattern causes defects in the Notch signalling pathway. Approximately 98% of cases are due to mutations or deletions at the locus causing haplo insufficiency of the JAG1 gene on chromosome 20p11.2-20p12; the remaining cases (~2%) are caused by mutations in NOTCH2. JAG1 and NOTCH2 are both components of the Notch signalling pathway,[8] which is a conserved cell signalling pathway that has a major role in embryonic development. ('Conserved' refers to the pathway relying on similar or identical sequences.)
Owing to the wide variety of clinical features and malformations of ALGS, and which are also seen in other syndromes and disorders, diagnosis is challenging. With advances in genetic testing, diagnosis can be made based on genotype; however, diagnosis usually involves both phenotypic and genotypic description. Phenotypic criteria have been updated in response to recent observations of abnormalities of the kidneys and vasculature (often in the head and neck). Three of the seven characteristic clinical criteria are sufficient for diagnosis, namely: cholestasis (without liver biopsy being required); ophthalmological abnormalities (commonly posterior embryotoxon); characteristic facial features (prominent forehead, deep-set eyes with moderate hypertelorism, pointed chin, and straight nose with a bulbous tip); cardiac defects (commonly peripheral pulmonary artery stenosis); skeletal abnormalities (commonly butterfly vertebrae); abnormalities of the kidneys; and vascular abnormalities.[9] Diagnosis can also be confirmed if there is a first-degree relative and two classic criteria are met.
Hepatic disease in patients with ALGS is highly variable, even when the same mutation is present. Hepatic disease is the highest cause of morbidity in ALGS and ranges from mild biochemical abnormalities to end-stage liver disease, which is seen in up to 15% of cases.[10] For patients who present with liver disease in infancy, 20 - 30% will develop intractable portal hypertension, cirrhosis or synthetic liver failure.[11] There is no guideline available to predict liver disease outcome in ALGS, although Mouzaki et al.[l2]recently reported that prediction of 'long-term hepatic outcomes in ALGS could be based on serum total bilirubin levels in the first two years of life, combined with the identification of fibrosis on liver biopsy performed in the first 5 years of life and the presence of xanthomata by age 5 years. In addition, in a cholestatic child with ALGS, an average total bilirubin (level) of above 3.8 mg/dL between the ages of 12 and 24 months represents a cut-off that by itself has significant predictive value for severe or mild hepatic outcomes in later life.' Early-onset cholestasis is predictive of a worse prognosis of liver disease. However, it is noted that severe liver complications are possible even after late onset of disease, requiring ongoing follow-up throughout life.[13]
Rare disease
A rare disease is defined as a condition that affects fewer than 200 000 people (USA) or less than 1 in 2 000 people (European Union).[14] With a prevalence of 1%, vWD is reported as the most common bleeding disorder. This rate includes all qualitative and quantitative forms of the disease, including patients who may be asymptomatic or have a very mild disease course. If those with a mild form of disease or who are asymptomatic are excluded, the prevalence is ~1 in 10 000. Therefore, bleeding forms of vWD can be considered a rare disease.[15] Men and women are stated to be affected equally and there is no evidence of racial or ethnic predisposition.
As mentioned, ALGS has an approximate prevalence of 1 in 30 000 people, and sometimes this rate is reported to be even less. ALGS therefore also qualifies as a rare disease. As for vWD, there is no evidence to suggest a racial, geographical or ethnic predisposition of this disorder.
The likelihood of these two disorders occurring in a single family is small. The diseases occur independently and the probability of simultaneous occurrence is simply the product of each event's individual probability. As vWD is congenital, the likelihood of offspring having vWD would range between 25% and 50% depending on the subtype. For people with ALGS, ~30 - 50% have an inherited gene mutation, while the remainder of cases develop spontaneously.[16] As ALGS is autosomal dominant, inheriting the gene mutation confers a 50% chance of phenotypic expression of the disorder. If our patient inherited ALGS, her likelihood of having both disorders would range between 12.5% and 25%. However, we know that our patient did not inherit ALGS, and therefore the likelihood is higher.
However, in a population, the chance of encountering a randomly selected patient with both ALGS and vWD is assessed differently. These disorders occur without gender, racial or ethnic predisposition and they are not found on the same or related chromosomal or genetic pathways. At a population level, the probability that such a patient exists ranges between 1 in 3 million and 1 in 300 million, depending on the severity of vWD. Therefore, the management of such a patient needs to be highly individualised.
An uncontrollable upper gastrointestinal bleed secondary to oesophageal varices is a potentially life-threatening situation in a patient with co-occurring vWD and ALGS, and there is also a risk of variceal development from increased portal hypertension in chronic liver disease. In our patient, this potential complication needs to be monitored and managed.
Between 15% and 30% of ALGS patients will develop end-stage liver disease with the risk of portal hypertension, but there is no definitive way to predict this outcome. Monitoring total serum bilirubin levels and the identification of liver fibrosis on biopsy during early childhood have predictive potential. In late childhood, as is the case with our patient, ongoing monitoring of liver function is part of management. In the case of our patient, an indication arose to exclude oesophageal varices owing to the development of splenomegaly and hypochromic anaemia.
Liver biopsy is the gold standard for the diagnosis of liver cirrhosis, but it does carry a bleeding risk. In their study of patients with vWD who underwent liver biopsy, Basu et al.[l7]found no episodes of major bleeding although 75% experienced minor local bleeding and ecchymosis, which resolved within 24 hours. However, the study did not include patients with type 3 vWD. In the case of our patient, other non-invasive techniques to monitor for liver cirrhosis are advised, such as magnetic resonance elastography or transient elastography. If a biopsy were indicated, DDAVP cover would decrease the potential bleeding risk.
The current gold standard for diagnosing oesophageal varices is oesophagogastroduodenoscopy. Other indicators, such as platelet count, spleen size, portal vein diameter, transient elastography, capsule endoscopy and measurement of portal pressure, may assist in diagnosis. As the direct measurement of portal pressure is invasive, the difference between wedged hepatic venous pressure and free hepatic venous pressure as an indication of the hepatic venous pressure gradient is considered a good predictor. However, the availability of this measurement is limited as it requires technical expertise and is costly. The technique also requires the insertion of a catheter at the antecubital, femoral or right jugular vein, which could pose a bleeding risk in a case such as ours.[18]
In patients who develop cirrhosis, about 50% develop varices, with an annual rate of variceal development of 5 - 15%. Endoscopic signs indicative of a high bleeding risk are a red wale sign and a medium or large varix. Once bleeding occurs in patients with cirrhosis, 'spontaneous cessation of bleeding occurs in only up to 40% of individuals, and the bleeding is associated with a mortality of 20% or more at 6 weeks';[19] there is also a high risk of rebleeding. The risk of cessation of bleeding from varices for our patient is likely to be significantly less than 40% and with the risk of rebleeding, long-term management would be required.
Although the combination of variceal bleeds and vWD is rare, there is a known gastrointestinal bleeding complication in vWD caused by angiodysplasia, which may require management of severe haemorrhage. The best results of pharmacological intervention are seen with plasma-derived vWF/FVIII concentrates administered in addition to red-cell blood transfusions and/or intravenous iron supplementation. Such replacement therapy is aimed at addressing generalised bleeding seen in angiodysplasia. In contrast, the standard approach to localised gastrointestinal bleeding is endoscopic, surgical or angiographic therapy.[20] Oesophageal varices are classified as localised bleeding sites and standard management of variceal haemorrhage needs to be available in light of the potential life-threatening outcome.
For patients with angiodysplasia, recurrent bleeding is a risk, and treatment with regular vWF/FVIII concentrates has been shown to provide the best prophylactic therapy. Long-term management of oesophageal varices aims to reduce portal pressure with a non-selective beta-blocker. The varices can also be obliterated through endoscopic ligation.
Conclusion
The likelihood of encountering a patient with co-occurring ALGS and vWD is extremely small. The management of such a patient could pose a risk of severe haemorrhage. Liver pathology is the greatest cause of morbidity in patients with ALGS. The potential risk of oesophageal variceal development owing to liver cirrhosis is a realistic concern and ongoing monitoring of the patient's liver function is required.
Should non-bleeding varices occur, prophylactic treatment should be implemented and the importance of including vWF/FVIII concentrates into standard treatment practice should be considered. If an acute bleed develops, emergency treatment must not be delayed. Standard oesophageal management must be employed along with the addition of vWF/FVIII concentrates.
Teaching points
The likelihood of encountering a patient with dual pathologies of ALGS and vWD is extremely small.
1. Liver pathology is the greatest cause of morbidity in patients with ALGS.
2. The potential risk of variceal development in a patient as described here warrants ongoing monitoring and follow-up.
3. The potential risk of life-threatening variceal bleeding is compounded by vWD.
4. Prophylactic treatment should be implemented, with the inclusion of vWF/FVIII concentrates being considered in standard treatment.
Declaration. None.
Acknowledgements. The authors thank both the patient and her mother for their consent to publishing this case report.
Author contributions. MJD conceptualised and designed the report, reviewed the literature and wrote the manuscript. BFJ and PW supervised the case analysis and reviewed the manuscript. All authors approved the final version of the manuscript.
Funding. None.
Conflicts of interest. None.
References
1. Ng C, Motto DG, Di Paola J. Diagnostic approach to von Willebrand disease. Blood 2015;125(13):2029-2037. https://doi.org/10.1182/blood-2014-08-528398 [ Links ]
2. Peyvandi F, Garagiola I, Baronciani L. Role of von Willebrand factor in the haemostasis. Blood Transfus 2011;9(Suppl 2):s3-s8. https://doi.org/10.2450/2011.002S [ Links ]
3. Leebeek FWG, Eikenboom JCJ. Von Willebrand's disease. N Engl J Med 2016;375(21):2067-2080. https://doi.org/10.1056/NEJMra1601561 [ Links ]
4. Pollak ES, Nagalla S. Von Willebrand disease workup. https://emedicine.medscape.com/article/206996-workup (accessed 1 November 2018). [ Links ]
5. Loran CS, Staros EB. Von Willebrand factor multimers. https://emedicine.medscape.com/article/2086389-overview (accessed 1 November 2018). [ Links ]
6. Schinco P, Castaman G, Coppola A, et al. Current challenges in the diagnosis and management of patients with inherited von Willebrand's disease in Italy: An Expert Meeting Report on the diagnosis and surgical and secondary long-term prophylaxis. Blood Transfus 2018;16(4):371-381. https://doi.org/10.2450/2017.0354-16 [ Links ]
7. Spinner NB, Gilbert MA, Loomes KM, Krantz ID. Alagille syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, eds. GeneReviews. Seattle: University of Washington, 1993 - 2020. [ Links ]
8. Turnpenny PD, Ellard S. Alagille syndrome: Pathogenesis, diagnosis and management. Eur J Hum Genet 2012;20:251-257. https://doi.org/10.1038/ejhg.2011.181 [ Links ]
9. Saleh M, Kamath BM, Chitayat D. Alagille syndrome: Clinical perspectives. Appl Clin Genet 2016;9:75-82. https://doi.org/10.2147/TACG.S86420 [ Links ]
10. Diaz-Frias J, Kondamudi NP. Alagille syndrome. In: Treasure Island. StatPearls Publishing, 2020. https://www.ncbi.nlm.nih.gov/books/NBK507827/ [ Links ]
11. Kamath BM, Munoz PS, Bab N, et al. A longitudinal study to identify laboratory predictors of liver disease outcome in Alagille syndrome. J Pediatr Gastr Nutr 2010;50(5):526-530. https://doi.org/10.1097/MPG.0b013e3181cea48d [ Links ]
12. Mouzaki M, Bass LM, Sokol RJ, et al. Early life predictive markers of liver disease outcome in an international, multicentre cohort of children with Alagille syndrome. Liver Int 2016;36(5):755-760. https://doi.org/10.1111/liv.12920 [ Links ]
13. Lykavieris P, Hadchouel M, Chardot C, Bernard O. Outcome of liver disease in children with Alagille syndrome: A study of 163 patients. Gut 2001;49(3):431-435. https://doi.org/10.1136/gut.49.3.431 [ Links ]
14. Genetic and Rare Diseases Information Center. FAQs about rare diseases. https://rarediseases.info.nih.gov/diseases/pages/31/faqs-about-rare-diseases (accessed 1 November 2018). [ Links ]
15. Veyradier A, Boisseau P, Fressinaud E, et al. A laboratory phenotype/genotype correlation of 1167 French patients from 670 families with von Willebrand disease: A new epidemiologic picture. Medicine 2016;95(11):e3038. https://doi.org/10.1097/MD.0000000000003038 [ Links ]
16. National Institute of Diabetes and Digestive and Kidney Diseases. Alagille syndrome. https://www.niddk.nih.gov/health-information/liver-disease/alagille-syndrome (accessed 1 November 2018). [ Links ]
17. Basu PP, Shah NJ, Aloysius MM, Rayapudi K, Brown R. A study on the safety of liver biopsy in patients with von Willebrand's disease. Open J Gastroenterol 2015;5(9):134-138. https://doi.org/10.4236/ojgas.2015.59022 [ Links ]
18. Suk KT. Hepatic venous pressure gradient: Clinical use in chronic liver disease. Clin Mol Hepatol 2014;20(1):6-14. https://doi.org/10.3350/cmh.2014.20.L6 [ Links ]
19. Maruyama H, Yokosuka O. Pathophysiology of portal hypertension and esophageal varices. Int J Hepatology 2012;2012:895787. https://doi.org/10.1155/2012/895787 [ Links ]
20. Franchini M, Mannucci PM. Gastrointestinal angiodysplasia and bleeding in von Willebrand disease. Thromb Haemost 2014;112(3):427-431. https://doi.org/10.1160/th13-11-0952 [ Links ]
21. Rosenberg HK, Markowitz RI, Kolberg H, Park C, Hubbard A, Bellah RD. Normal splenic size in infants and children: Sonographic measurements. Am J Roentgenol 1991;157(1):119-121. https://doi.org/10.2214/ajr.157.L2048509 [ Links ]
 Correspondence:
Correspondence:
M J Dempster
dmpmeg001@yahoo.com
Accepted 10 December 2020

