










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Child Health
On-line version ISSN 1999-7671
Print version ISSN 1994-3032
S. Afr. j. child health vol.15 n.1 Pretoria Apr. 2021
http://dx.doi.org/10.7196/sajch.2021.v15i1.1767
CASE REPORT
Abetalipoproteinaemia arising from a new variant of microsomal triglyceride transfer protein in a child presenting with chronic fat malabsorption
C B EkeI; A D MaraisII; R J De LacyIII; A J HooperIV; B D RatanjeeV; F A GoddardVI
IFWACP, M Phil; Division of Gastroenterology, Department of Paediatrics and Child Health, Red Cross War Memorial Childrens Hospital and University of Cape Town, South Africa; and Department of Paediatrics, College of Medicine, University of Nigeria, Enugu, Nigeria
IIFCP (SA); Division of Chemical Pathology, Department of Pathology, University of Cape Town, South Africa
IIIFCPaed (SA); Division of Gastroenterology, Department of Paediatrics and Child Health, Red Cross War Memorial Childrens Hospital and University of Cape Town, South Africa
IVPhD; Department ofClinical Biochemistry, PathWest Laboratory Medicine WA, Fiona Stanley Hospital Network, Perth, Australia
VBSc; Lipidology Division, Department of Internal Medicine, Groote Schuur Hospital, Cape Town, South Africa
VIPhD; Division of Gastroenterology, Department of Paediatrics and Child Health, Red Cross War Memorial Childrens Hospital and University of Cape Town, South Africa
ABSTRACT
This case report describes a patient who failed to thrive and lagged in development in the first year of her life and did not grow as expected. Though a history of diarrhoea was present, stool analysis was not conclusive for malabsorption of fat. Fat malabsorption may be due to various disorders of exocrine function of the pancreas as well as problems in the intestine. Abetalipoproteinaemia is a rare inherited condition in which neither the intestine nor the liver can secrete lipoproteins. This diagnosis was made on lipoprotein investigation by demonstrating the absence of lipoproteins containing apolipoprotein B (apoB). Lipoprotein investigation is generally normal in the parents of the recessively inherited condition of microsomal triglyceride transfer protein (MTTP) deficiency but in homozygous hypobetalipoproteinaemia owing to mutations in apoB that also impair lipoprotein assembly, the parents have low cholesterol concentrations. Patients with abetalipoproteinaemia improve markedly on dietary fat restriction but may benefit from medium-chain triglycerides as part of supplying their energy needs and, importantly, require high doses of vitamin F as well as other fat-soluble vitamins, to avoid neuromuscular degeneration and retinitis pigmentosa.
Microsomal triglyceride transfer protein (MTTP) is a 97 kDa protein encoded by the MTTP gene in chromosome 4q22-24. It forms a heterodimer with protein disulphide isomerase to transfer neutral lipid to newly synthesised apolipoprotein B (apoB) in the rough endoplasmic reticulum. Dysfunction in MTTP therefore results in impaired apoB processing for secretion and abrogates transport of lipids and fat-soluble vitamins usually carried within lipoprotein particles.[1-6]
Impaired transport of MTTP, either as homozygous or compound heterozygous mutations in MTTP, causes abetalipoproteinaemia (ABL), also known as Bassen-Kornzweig syndrome. This autosomal recessive disorder is characterised by complete absence of apoB-containing lipoprotein in the blood plasma. This may be indistinguishable in its clinical presentation from homozygous hypobetalipoproteinaemia owing to truncations of apoB.[3-5] Both causes of ABL are from a complete failure of assembling lipoproteins in the intestine and the liver, resulting in lipid malabsorption and impaired transport of triglyceride and fat-soluble vitamins, especially for vitamin E.
ABL is extremely rare, with fewer than 100 cases reported in the literature and has an estimated frequency of about 1 in a million.[1-4] A child with ABL may present with failure to thrive in infancy, disturbances in neuromuscular function as well as ophthalmic complications, particularly an atypical retinitis pigmentosa resulting from vitamin F and A deficiencies.[5]
In ABL, both enterocytes and hepatocytes accumulate lipid droplets as a result of impaired export of lipid in lipoproteins.[5,7-10] Failure to assemble chylomicrons causes fat malabsorption, which not only results in impaired energy supply to the growing neonate and infant but also impairs the transport of vitamin F that protects against lipid peroxidation. Deficiency of vitamin F can harm especially retinal and neuromuscular function. The age of presentation is variable but the majority of patients present in infancy following introduction of fatty diets.[3,5,11]
No definitive therapy is currently available for ABL. Nutritional modification involving a low-fat diet as well as supplementations of essential fatty acids and high doses of fat-soluble vitamins (ADEK) have been shown to ameliorate the condition, and to improve growth and overall quality of life in affected children.[4-6] Importantly, the diet should supply adequate energy particularly in growing children.
In this report, we describe the clinical presentation, laboratory findings and successful treatment of a child who presented with the features of ABL found to be due to a novel homozygous loss-of-function MTTP variant. The low plasma cholesterol was proven to be due to the absence of all apoB-containing lipoproteins by lipoprotein electrophoresis.
Case
An index child, female, presented initially at age 5 months with poor weight gain, recurrent episodes of frequent loose stools together with abdominal distention and developmental delay. She was the second child of consanguineous parents of Palestinian descent. There was no family history of similar illnesses. No problems were noted during pregnancy. She weighed 3.07 kg at birth. At presentation she weighed 5.4 kg (weight-for-age z-score, WAZ <-3) and had a length of 65 cm (length-for-age z-score, LAZ -1). She had frequent loose stools (about 6-8 times/day) which intermittently had the features of steatorrhoea (described as greasy stools and floating on water).
Physical examination revealed a thin female child with obvious developmental delay (poor holding of her head at age 6 months, she sat at 9 months, and stood with support at 18 months), generalised low muscle bulk, distended abdomen, mild hepatosplenomegaly, and normal visual acuity and neurologic examination. No abnormality was found on retinal fundoscopy.
Laboratory investigations revealed a microcytic anaemia-haemoglobin level of 10 g/dL (range 11 - 13 g/dL), and mean corpuscular volume of 62.9 fl. Peripheral blood smear revealed acanthocytosis of the red blood cells.
Faecal fat histology showed scanty faecal fat (2%) based on the method proposed by Khouri et al., [12] with a normal stool alpha I-antitrypsin level. Coeliac disease screening, sweat testing for cystic fibrosis, blood glucose, immunoglobulins (IgA, IgG, IgM), retroviral polymerase chain reaction and haemoglobin electrophoresis did not reveal any abnormal results. The karyotype was normal (46XX).
The serum transaminase activities were mildly deranged: alanine transaminase 95 IU/L (range 5-45 iu/L) and aspartate transaminase 85 IU/L (range 15 - 40 IU/L), respectively.
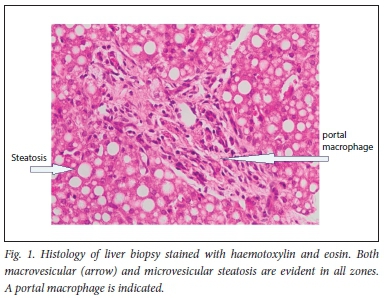
Abdominal ultrasound examination showed mild hepatosplenomegaly. Liver histology revealed diffuse macrovesicular and microvesicular steatosis in all zones (Fig. 1).
The serum vitamin A level was 0.65 (0.70 - 1.50) μmοΙ/L, vitamin D level 44.9 nmol/L (50 - 70 nmol/L), serum vitamin F 0.41 (7.0 - 21.0) μmοΙ/L, and international normalised ratio (INR) was 0.93 at presentation.
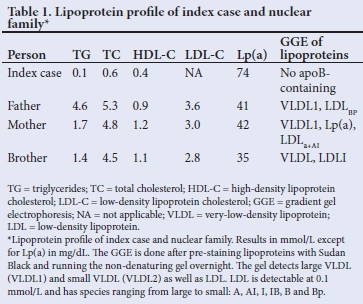
The fasting lipid profile was of severe hypocholesterolaemia: total cholesterol 0.79 mmol/1, high-density lipoprotein cholesterol (HDL-C) 0.54 mmol/L, low-density lipoprotein cholesterol (LDL-C) 0.06 mmol/L, and triglyceride (TG) was 0.01 mmol/L. The lipid profile of the father showed mild hypertriglyceridaemia but his other lipid parameters were within normal limits. The mother and brother had normal lipid profiles as shown in Table 1.
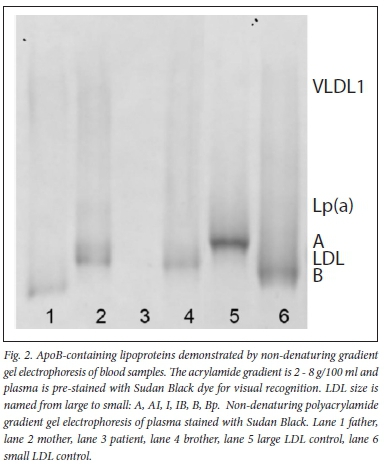
The apoB level was less than 0.20 g/L while the gradient gel electrophoresis (GGE) of lipoproteins did not display apoB-containing lipoproteins in the index case (see Table 1, and lane 3 in Fig. 2).
Massively parallel sequencing of lipid genes with targeted analysis of monogenic hypocholesterolaemic genes (APOB, MTTP, PCSK9, SAR1B, ANGPTL3) revealed that the proband is homozygous for a new pathogenic variant of MTTP gene, c. 1342 1344+ldelAAAG, consistent with a diagnosis of ABL. The variant is predicted to cause defective splicing, as it abolishes the MTTP intron 11 splice donor site. Oesophagogastroduodenoscopy with multiple small intestinal biopsies was planned but deferred following receipt of the genetic result.
The patient commenced a low-fat diet with the inclusion of medium-chain triglyceride (MCT) for energy balance. Essential fatty acids and high doses of fat-soluble vitamins were initially started, using the recommendations proposed by Lee and Hegele,[5] as follows: vitamin A 2 000 IU/day, vitamin D 1 200 IU/day, vitamin F 600 IU/day and vitamin K 5 mg/day. Oral iron, folate, zinc and multivitamin supplements were also started Doses of the fat-soluble vitamins including vitamin F were adjusted accordingly based on the weight measurements at follow-up visits and serial plasma vitamin levels assays.
The patient improved remarkably, with resolution of all symptoms including chronic diarrhoea and steatorrhoea. Her developmental delay was reversed and liver enzymes normalised. Her growth has continued to improve since on treatment: at the age of 4 years, 8 months, she weighed 20.6 kg (z-score progressed from +1 to +2 while her height was 114 cm (z-score progressed from -1 to 0). Her serum vitamin F level still runs at lower limits, 3.1 (7.0 - 21.0) μmοl/L despite being on high-dose vitamin F supplementation. Vitamin A concentration remained low (0.41 μmοl/L) but vitamin D (60.9 ng/ml) and INR (INR 1.18, prothrombin time (test) 13.9 seconds, prothrombin time (control) 12.0 seconds), were normal. She continues to be followed up on an outpatient basis.
Discussion
This report describes a novel 4 nucleotide deletion in MTTP (variant: c. 1342_1344+1 delAAAG) causing ABL with malabsorption. The diagnosis was suggested by the clinical constellation of apparent autosomal recessive inheritance, steatorrhoea and failure to thrive, with very low plasma cholesterol concentration and the absence of apoB-containing lipoproteins.
When there is a history of chronic passage of greasy, frothy voluminous stool alongside weight loss or poor growth in a child, efforts should be made to identify the cause of fat malabsorption. Commonly the causes of fat malabsorption will include biliary atresia, exocrine pancreatic insufficiency including cystic fibrosis, decreased enterohepatic circulation of bile acids, or excessive deconjugation of bile acids by bacterial overgrowth.[13]
The first step should be faecal fat analysis to identify the fat in faeces. The spot stool specimen can be helpful but the gold standard is still the 3-day stool collection for fat estimation when dietary fat intake is strictly calculated by a nutritionist. However, this process is very cumbersome and the spot faecal fat histology in places where it is obtainable is handy and very useful. In general, more than 6% faecal fat excretion is considered abnormal.[13] The index patient had scanty faecal fat of 2%, and hence the stool fat analysis was inconclusive for fat malabsorption. The diagnosis of ABL was, however, made with the lipoprotein investigation by demonstrating the absence of lipoproteins containing apoB.
The lipid profiles of the parents were useful in excluding homozygous hypobetalipoproteinaemia as neither obligate heterozygous parent had half-normal levels of apoB-containing lipoproteins.[1,2,5,12]Similarly, undetectable concentrations of plasma apoB and LDL in the index case excluded chylomicron retention disease, another autosomal recessive disorder that results in fat malabsorption, but this disorder sustains normal (hepatic) apoB-containing lipoproteins; thus the final diagnosis is ABL.[1,14] The routine laboratory measurement of apoB did not indicate complete absence of apoB. Lipoprotein electrophoresis can further assist in describing the presence and type of lipoproteins in both hyperlipidaemias and hypolipidaemias.
Lipoprotein investigation by gradient gel electrophoresis indicated that the index case had no demonstrable apoB-containing lipoproteins. The father had prominent large very-low-density lipoprotein (VLDL), explaining the hypertriglyceridaemia, and the brother's sample contained VLDL and LDL; and thus, along with his lipid profile, the diagnosis of ABL was excluded. The father, mother and brother had varying levels of LDL. It is interesting to note that the index case, in whom the highest concentration of Lp(a) was reported, did not display a distinct lipoprotein species at the expected size, unlike her mother, whose Lp(a) was visible on electrophoresis (Fig. 2). It is suggested that in the index case, the assay detected apo(a) that is not associated with LDL to which it is normally bound covalently. The absence of this band in the father who had a similar Lp(a) concentration as the mother, may be due to the presence of a more diffuse range of Lp(a) commensurate with hypertriglyceridaemia.[15] Lipoprotein analysis could be meaningful in predicting the parents' cardiovascular risk as well.
Although there is no cure for this condition, judicious nutritional modification may retard the progression or ameliorate the symptoms of ABL.[1-3,5] The patient consumed a very low-fat diet along with high doses of fat-soluble vitamins and essential fatty acids supplementation. The MCT-based feeds were initially used to correct her energy malnutrition. The fatty acids from MCT are directly transported in the portal vein and bypass chylomicron assembly, with excellent tolerance and outcomes.[3] Supplementations of essential fatty acids including linoleic and linolenic acids prevent the development of essential fatty acids deficiency.
Fat-soluble vitamins supplementation is essential to prevent several complications of ABL including retinitis pigmentosa, an ophthalmological complication owing to deficiency of vitamin E. Despite high-dose supplementation of vitamin E, however, the plasma levels of vitamin F did not return to normal because vitamin F is usually transported in circulating lipoproteins.[14,16] Red blood cell or tissue levels of vitamin F may be more informative about her vitamin F status but are not performed in the local laboratory.[16] Adjustments of plasma tocopherol concentration (vitamin E) for total lipid (cholesterol), or even to cholesterol, cholesterol + triglyceride, cholesterol + triglyceride + phospholipid and even in isolated lipoprotein fractions may improve assessment of tocopherol status in the setting of hyperlipidaemia but are not reliable for ABL. When all lipoproteins containing apoB are absent, the plasma tocopherol value is practically that of tocopherol in HDL.
It is interesting to note that developmental delay as well as impaired growth improved with strict nutritional therapy in the index patient, fortunately before developing further neurological complications.
Conclusion
ABL is a rare autosomal recessive genetic disorder resulting in fat malabsorption. Nevertheless, this diagnosis should be considered with a high index of clinical suspicion in any child with poor growth or weight loss and chronic diarrhoea or steatorrhoea, especially in the setting of consanguineous parentage.
A similar problem of homozygous hypobetalipoproteinaemia may cause ABL and will be suggested by low cholesterol levels in both parents. At later stages of life, neuromuscular complications such as retinitis pigmentosa and ataxia may set in but can be prevented by early nutritional therapy including high doses of fat-soluble vitamins and essential fatty acids supplementation.
Declaration. None.
Acknowledgements. We acknowledge the index patient and her family for agreeing to complete investigation and anonymous teaching. We are grateful to Ms Lan Nguyen for her assistance with DNA sequencing.
Author contributions. ADM, EAG, RJD, CBE: Conceptualised the study. CBE: Initial manuscript draft. CBE, ADM, AJH, RJD, BDR, EAG: Critical editing of the manuscript for important intellectual content. All authors approved the final draft.
Funding. None.
Conflicts of interest. None.
References
1. Rashtrain P, Sani MN, Jalilian R. A male infant with abetalipoproteinaemia: A case report from Iran. Middle East J Dig Dis 2015;7:181-184. [ Links ]
2. Zamel R, Khan R, Pollex RL, et al. Abetalipoproteinaemia: Two case reports and literature review. Orphanet J Rare Dis 2008;3:19. https://doi.org/10.1186/1750-1172-3-19 [ Links ]
3. Davidson MO, Levy E. Congenital disorders of lipid transport. In: Guanda-lini S, Dhawan A, Branski D, editors. Textbook of Pediatric Gastroenterology, Hepatology and Nutrition : A Comprehensive Guide to Practice. Cham: Springer International, 2016:437-444. [ Links ]
4. Burnett JR, Hooper AJ, Hegele RA. Abetalipoproteinemia. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews (R). Seattle: University of Washington; 1993-2019. [ Links ]
5. Lee J, Hegele RA. Abetalipoproteinaemia and heterozygous hypobetalipopro-teinaemia: A framework for diagnosis and management. J Inherit Met Dis 2014;37(3):333-339. https://doi.org/10.1007/sl0545-013-9665-4 [ Links ]
6. Welty FK. Hypobetalipoproteinaemia and abetalipoproteinaemia. Curr Opin Lipidol 2014;25(3):161-168. https://doi.org/10.1097%2FMOL.0000000000000072 [ Links ]
7. Whitfield AJ, Barrett HR, Van Bockxmeer FM, et al. Lipid disorders and mutations in the APO B gene. Clin Chem 2004;50(10):1725-1732. https://doi.org/10.1373/clinchem.2004.038026 [ Links ]
8. Di Fillippo M, Crehalet H, Samson-Bouma ME, et al. Molecular and functional analysis of two new mutations in an atypical case of abetaliopoproteinaemia. J Lipid Res 2012;53(3):548-555. https://doi.org/10.1194/jlr.m020024 [ Links ]
9. Uslu N, Gurakan F, Yuce A, et al. Abetalipoproteinaemia in an infant with severe clinical phenotype and a novel mutation. Turk J Pediatr 2010;52(1):73-77. [ Links ]
10. Wang J, Hegele RA. Microsomal triglyceride transfer protein (MTP) gene mutation in Canadian subject with abetalipoproteinaemia. Hum Mutat 2000;15(3):294-295. https://doi.org/10.1002/(sici)10981004(200003)15:3%3C294::aid-humu14%3E3.0.co;2-e [ Links ]
11. Berriort-Raroqueaux N, Aggerbeck LP, Samson-Bouma M, et al. The role of the microsomal triglyceride transfer protein in abetalipoproteinaemia. Ann Rev Nutr 2000;20:663-697. https://doi.org/10.1146/annurev.nutr.20.l.663 [ Links ]
12. Khouri MR, Huang G, Shiau YE Sudan stain of fecal fat: New insight into an old test. Gastroenterology 1989:96:421-427. https://doi.org/10.1016/0016-5085(89)91566-7 [ Links ]
13. Gupta R. Diarrhea. In: Wyllie R, Hyams JS, Kay M, editors. Pediatric gastrointestinal and liver disease. 5th ed. New York: Elsevier; 2016:114. [ Links ]
14. Cureq C, Rester L, Drai J, et al. Establishment of reference values of alpha-tocopherol in plasma, red blood cells and adipose tissue in healthy children to improve the management of chylomicron retention disease, a rare genetic hypocholesterolaemia. Orphanet J Rare Dis 2016;11(1):14. https://doi.org/10.1186/sl3023-016-0498-8 [ Links ]
15. Bach-Ngohou K, Giraud F, Krempf M, et al. Influence of remnant accumulation markers on plasma concentrations of two lipoprotein(a) subspecies (containing or free of apoE). Metabolism 2001;50(3):277-282. https://doi.org/10.1053/meta.2001.21031 [ Links ]
16. Segal S, Sharma S. Ophtha-problem. Vitamin A and vitamin E. Can Fam Physician 2005;51(8):85-86. [ Links ]
 Correspondence:
Correspondence:
CBEke
christopher.eke@unn.edu.ng
Accepted 27 August 2020

