










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Child Health
versão On-line ISSN 1999-7671
versão impressa ISSN 1994-3032
S. Afr. j. child health vol.15 no.1 Pretoria Abr. 2021
http://dx.doi.org/10.7196/sajch.2021.v15i1.1731
CASE REPORT
Multifocal kaposiform haemangioendothelioma: Another sirolimus success story
B LackhooI; B RoweII
IDCH (SA), Dip HIV Man (SA); Department of Paediatrics, Chris Hani Baragwanath Academic Hospital, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIMMed (Paed); Department of Paediatrics, Chris Hani Baragwanath Academic Hospital, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
Kaposiform haemangioendothelioma (KHE) is a rare, infiltrative, vascular tumour of infancy and early childhood that is often complicated by the Kasabach-Merritt phenomenon (KMP), a consumptive coagulopathy. We describe the case of a 7-year-old boy who presented with a left shoulder mass, splenic hypodensities, bony lesions, and a coagulopathy. The histopathological findings were consistent with KHE. Emergent treatment of KHE involves correction of the coagulopathy with blood products, while pharmacotherapy is aimed at reducing the tumour size. Emerging data suggest a role for mammalian target of rapamycin (mTOR)-inhibitors, e.g. sirolimus, in the treatment of progressive KHE, to which our patient had an excellent clinical response. Our recommendation is that KHE should be considered in the differential diagnoses of an ill-defined, infiltrating, soft-tissue mass. Pharmacotherapy including an mTOR-inhibitor should be instituted in patients with symptomatic KHE. Long-term follow-up of residual KHE lesions is essential as they follow an unpredictable course.
Kaposiform haemangioendothelioma (KHE) is a rare, infiltrative, vascular tumour predominantly occurring in early childhood.[1] KHE was first described by Zuckerberg in 1993 and distinguished from the more common benign haemangioma.[1] Rare cases of multifocal KHE have been reported,[2] and we add to this series by describing a patient who presented with KHE involving the soft tissues, spleen and bones.
Case
A 7-year-old boy presented with a history of assault involving the left shoulder. He was given analgesia and discharged. A month later he re-presented with shortness of breath, cough and worsening left shoulder pain. A left tension haemothorax was diagnosed and an intercostal drain inserted. Fractures of the left scapula and left clavicle were noted and were of concern in terms of pathological fractures. A computed tomography angiogram (CTA) was performed by the referral hospital to investigate for an active bleed as his haemoglobin level continued to decrease despite transfusion. It showed active bleeding from the left subclavian and supreme intercostal vein. He was referred to tertiary care.
Upon arrival at our institution, he was pale and noted to have an extensive violaceous swelling over the left neck and shoulder of a boggy quality; the swelling was extremely painful. He was in respiratory distress with decreased breath sounds on the left.
He underwent a repeat CTA which showed a left supraclavicular, ill-defined hypodense mass with extension into the superior mediastinum with lytic bony lesions in the left clavicle and left scapula. A massive haematoma involving the left shoulder and left lateral chest wall with a large left pleural effusion was present. No vascular injury was demonstrated. Multiple lytic vertebral body lesions and splenic hypodensities were present. His full blood count showed a bicytopenia with a haemoglobin level of 7.0 g/dL and a platelet count of 10 χ 109/L. A limited baseline coagulation screen showed an international normalised ratio (INR) of 1.4 and a partial thromboplastin time (PTT) of 28.3 sec - he had already been transfused with fresh frozen plasma prior to arrival. Bone marrow examination showed no malignant infiltration.
He required multiple transfusions of red cells and platelets in the first 4 days of admission. He underwent a biopsy of the clavicle and humeral head, and histopathological examination showed a vascular neoplasm with widespread necrosis, haemorrhage and focal areas of tumour cells that displayed epithelioid features and slit-like morphology. Mitotic activity was sparse with an average count of 0/10 high power field (HPF). The tumour cells stained diffusely positive for CD31, CD34 and D2-40. Of note, HHV8, CD1a, CD99, S100, CD56, AE1/AE3, CAM5.2, EMA, CD68, and desmin and myogenin stains were negative. A final diagnosis of a left extremity KHE with consumptive coagulopathy, Kasabach-Merritt phenomenon (KMP), was made. The vertebral body lytic lesions and splenic hypodensities represented multifocal haemangioendotheliomas.
Treatment was initiated with weekly vincristine, and daily prednisone and sirolimus. He completed 8 weeks' therapy with vincristine, during which time the prednisone was tapered and stopped.
He had a follow-up CT scan after 8 weeks of chemotherapy, which showed a fat tissue density in the head of the left humerus and coracoid process of the scapula with no mass-like lesions. The haemothorax had resolved completely. The splenic hypodense lesions had similarly resolved. He remains on sirolimus as monotherapy and will have follow-up imaging at 4-monthly intervals.
Discussion
KHE is categorised as a vascular tumour of intermediate malignancy owing to its locally aggressive nature.[3] The mortality rate can be as high as 30% when complicated by KMP, which develops in 70% of cases.[1] Compression and infiltration of vital structures add to the morbidity of KHE as it infiltrates across multiple tissue planes.[3]
KHE most often presents as a tumour located superficially in the skin or infiltrating deeper into the soft tissues.[1] It has ill-defined margins with an erythematous or violaceous hue.[1] In a retrospective review of 231 children with KHE, Schmid et al.[4] described the common sites for infants and toddlers as the head and neck (18%), trunk (29%) and extremities (25%). In older children, the tumours were more likely to present in the extremities.[4] Local trauma may trigger episodic engorgement and painful expansion of the lesion.[5] Consistent with the literature, our patient's KHE lesion increased in size and tenderness following trauma to the shoulder. Approximately 10% of KHEs occur in extracutaneous locations, notably in the retroperitoneum, mediastinum and bones.[4] Regional lymph node metastases have been documented but no distant metastatic disease has been reported to date.[5]
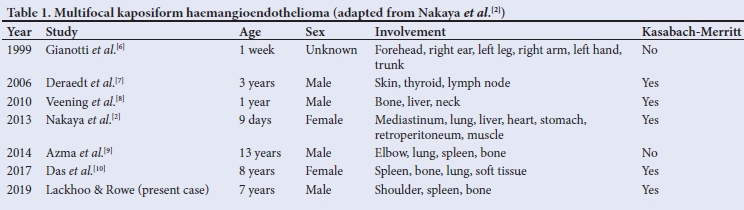
Multifocal KHE has been reported in the literature (Table 1).[2,6-10]. There is a matter of debate as to whether it should be considered a multifocality or metastasis because of the separate locations, but no clear histological features of malignancy have been described in the documented cases,[2] nor in our patient's histology.
KMP, a life-threatening coagulopathy, is associated exclusively with KHE[3] and is not a feature of benign haemangiomas. Clinically KMP is characterised by a painful, rapidly enlarging, engorged tumour.[5] It is associated with severe thrombocytopenia, hypofibrinogenaemia, elevated markers of coagulation and various degrees of anaemia.[5] The coagulopathy occurs secondary to intralesional platelet trapping that activates the coagulation cascade with secondary consumption of clotting factors and bleeding.[5]
The radiological modality of choice to evaluate KHE is magnetic resonance imaging (MRI), which may aid in differentiating KHE from other tumours.[5,11] The characteristic MRI features of KHE are masses that exhibit an infiltrative pattern, mild hyperintensity on T2WI, heterogeneous enhancement and destruction/remodelling of adjacent bone.[11] This is in contrast to benign haemangiomas which present as well-defined lesions with marked hyperintensity on T2WI, homogeneous enhancement and without overt destruction of bone.[5]
KHE lesions have both a vascular and lymphatic component as tumour cells express the lymphatic markers D2-40 and PROX-1. KHE lesions are characterised by irregular nodules that develop in an infiltrative pattern.[3,5] Slit-like apertures in the vessels are similar to the histological findings of Kaposi sarcoma but human herpes virus-8 transcripts are uniformly absent.[3] KHE tumour nodules contain vessels that are well canalised resembling capillary haemangioma, while other vessels are poorly canalised, and appear solid; this is a distinctive histological feature of KHE.[3,5] On immunohistochemical staining, endothelial cells express CD31 and CD34.[1,3] Negative glucose transporter isoform 1 (GLUT-1) immunostaining differentiates KHE from benign haemangiomas.[3]
The unique architecture of KHE may promote turbulence of blood flow leading to platelet aggregation, trapping and coagulopathy.[5]
Spontaneous involution of KHE lesions is rare.[11] This is in contrast with benign haemangiomas which inevitably involute over time after a proliferative phase in infancy. Complete surgical resection is the gold standard of cure of KHE, but total resection is often not feasible.[5,11] The aim of pharmacological management is not necessarily to cure but to decrease the tumour size and correct the coagulopathy.[5] Acute management of the coagulopathy includes transfusion with fresh frozen plasma and cryoprecipitate[11] Platelet transfusion may potentiate an increase in tumour size and should be reserved for patients with active bleeding.[5] Because of the rarity of KHE, data on treatment options remain limited to case series and reports. Previous therapies include propranolol, chemotherapy, aspirin, steroids, radiotherapy, interferon alpha and embolisation, as single or combination therapies, each with varying results.[4] Propranolol has been successful in the treatment of infantile haemangiomas but has little or no benefit in the treatment of KHE.[4,5] Combination chemotherapy with vincristine, actinomycin D and cyclophosphamide has reportedly reduced KHE tumour size; however, it may cause serious complications. Interferon alpha has been associated with spastic diplegia.[5] According to a 2013 multispecialty expert panel, the consensus recommendation for KHE associated with KMP was intravenous vincristine and oral prednisolone.[5] Patients with enlarging, symptomatic KHE that requires treatment, but without KMP, should have oral prednisolone at 2 mg/kg/d.[5] Tumours that are asymptomatic or not involving a vital organ may be observed. Since the publication of the consensus guidelines,[5] numerous case reports and a multicentre retrospective cohort study by Ji et al.[12] have confirmed sirolimus, a mammalian target of rapamycin (mTOR)-inhibitor, to be very effective in the treatment of progressive KHE. Cutaneous vascular malformations were found to overexpress mTOR.[12] Sirolimus has since surpassed vincristine and steroids as the treatment of choice in KHE. Sirolimus is a well-tolerated oral agent;[11] a potential drawback is the recommendation to monitor drug levels, as a trough level of 10 - 15 ng/mL is advised.[11] Potential side-effects of sirolimus include mucositis, angio-oedema, hyperlipidaemia, hypercholesterolaemia and rarely pneumonitis.[13] Sirolimus drug level monitoring is not available in the public health care setting in South Africa. Thorough clinical examination and monitoring of biochemical parameters, including lipid levels, should be used as surrogate markers for potential toxicity. A 2018 case report documents the death of two infants who received sirolimus as treatment for their KHE.[14] The authors postulate that the infants developed a sirolimus-related pneumonia. This pneumonia with atypical features (no fever, normal white cell count) was attributed to a combination of the immature immune system of the infants as well as the immunosuppression caused by sirolimus and steroids.[14] The authors recommend the use of antibiotic prophylaxis with sirolimus, although this is not yet an accepted recommendation. The optimum treatment duration with sirolimus is unknown, but the most dramatic treatment responses were shown to occur in the first 6 months of therapy.[11]
In our patient, the initial presentation of KHE with KMP was dismissed as a soft-tissue injury, leading to a delay in diagnosis. He had an excellent response to the combination therapy of prednisone, vincristine and sirolimus. The pharmacological cure of KHE is neither complete nor permanent, and any residual lesions may remain in a semi-dormant state and follow an unpredictable disease course necessitating long-term follow-up.
Conclusions
KHE should be considered in the differential diagnosis of an ill-defined, infiltrating soft-tissue mass, and early consideration should be given to imaging of the lesion, preferably by MRI. Timeous diagnosis and early institution of management can ameliorate the often-associated KMP with KHE and decrease tumour size and its compressive effects. Sirolimus is an encouraging treatment option because of its low side-effect profile and the added benefit of an oral preparation. Our experience with sirolimus echoes previous success documented in other case series and reports. We suggest long-term follow-up of residual KHE lesions to monitor for chronic pain, functional impairment and recurrence.
References
1. Croteau SE, Liang MG, Kozakewich HP, et al. Kaposiform hemangioendothelioma: Atypical features and risks of Kasabach-Merritt phenomenon in 107 referrals. J Pediatr 2013;162(1):142-147. https://doi.org/10.1016/j.peds.2012.06.044 [ Links ]
2. Nakaya T, Morita K, Kurata A, et al. Multifocal kaposiform hemangioendothelioma in multiple visceral organs: An autopsy of 9-day-old female baby. Hum Pathol 2014;45(8):1773-1777. https://doi.org/10.1016/j.humpath.2014.03.019 [ Links ]
3. Lyons LL, North PE, Lai FM, et al. Kaposiform hemangioendothelioma: A study of 33 cases emphasizing its pathologic, immunophenotypic, and biologic uniqueness from juvenile hemangioma. Am J Surg Pathol 2004;28(5):559-568. https://doi.org/10.1097/00000478-200405000-00001 [ Links ]
4. Schmid I, Klenk AK, Sparber-Sauer M, Koscielniak E, Maxwell R, Haberle B. Kaposiform hemangioendothelioma in children: A benign vascular tumour with multiple treatment options. World J Pediatr 2018;14(4):322-329. https://doi.org/10.1007/s12519-018-0171-5 [ Links ]
5. Drolet BA, Trenor CC, Brandao LR, et al. Consensus-derived practice standards plan for complicated kaposiform hemangioendothelioma. J Pediatr 2013;163(1):285-291. https://doi.org/10.1016/j.peds.2013.03.080 [ Links ]
6. Gianotti R, Gelmetti C, Alessi E. Congenital cutaneous multifocal Kaposiform hemangioendothelioma. Am J Dermatopathol 1999;21:557-561. https://doi.org/10.1097/00000372-199912000-00010 [ Links ]
7. Deraedt K, vander Poorten V, van Geet C, Renard M, de Wever I, Sciot R. Multifocal kaposiform haemangioendothelioma. Virchows Arch 2006; 448:843-846. https://doi.org/10.1007/s00428-006-0177-6 [ Links ]
8. Veening MA, Verbeke JI, Witbreuk MM, Kaspers GJ. Kaposiform (spindle cell) hemangioendothelioma in a child with an unusal presentation. J Pediatr Hematol Oncol 2010; 32:240-242. https://doi.org/10.1097/mph.0b013e3181ced284 [ Links ]
9. Azma R, Alavi S, Khoddami M, Arzanian M, Nourmohommad A, Esteghamati S. Multifocal kaposiform hemangioendothelioma of soft tissue with bilateral pulmonary involvement in an adolescent. Korean J Pediatr 2014;57(1):500-504. https://doi.org/10.3345/kjp.2014.57.11.500 [ Links ]
10. Das A, Bansal D, Chatterjee D, Das A. Kaposiform hemangioendothelioma: Multifocal involvement, chylothorax, and Kasabach-Merritt phenomenon. J Pediatr Hematol Oncol 2017;39(2):153-154. https://doi.org/10.1097/MPH.0000000000000700 [ Links ]
11. Ryu YJ, Choi YH, Cheon J, et al. Imaging findings of kaposiform hemangioendothelioma in children. Eur J Radiol 2017;86(1):198-205. https://doi.org/10.1016/j.ejrad.2016.11.015 [ Links ]
12. Ji Y, Chen S, Xiang B, et al. Sirolimus for the treatment of progressive kaposiform hemangioendothelioma: A multicenter retrospective study. Int J Cancer 2017;141(4):848-855. https://doi.org/10.1002/ijc.30775 [ Links ]
13. Blatt J, Stavas J, Moats-Staats B, Woosley J, Morrell DS. Treatment of childhood Kaposiform haemangioendothelioma with sirolimus. Pediatric Blood Cancer 2010;55(7):1396-1398. https://doi.org/10.1002/pbc.22766 [ Links ]
14. Ying H, Qiao C, Yang X, Lin X. A case report of 2 sirolimus-related deaths among infants with kaposiform hemangioendothelioma. Pediatrics 2018;141(5):S425-S429. https://doi.org/10.1542/peds.2016-2919 [ Links ]
 Correspondence:
Correspondence:
B Lackhoo
bhavisha.l@hotmail.com
Accepted 27 August 2020


{kind=link}