










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Child Health
versão On-line ISSN 1999-7671
versão impressa ISSN 1994-3032
S. Afr. j. child health vol.13 no.4 Pretoria Dez. 2019
http://dx.doi.org/10.7196/sajch.2019.v13i4.1558
RESEARCH
Fanconi anaemia: A perspective on phenotype, investigations and outcomes from central South Africa
D K Stones
MB ChB, FCPaed, MMed (Paed), DCH; Department of Paediatrics and Child Health, Faculty of Health Sciences, University of the Free State, Bloemfontein, South Africa
ABSTRACT
BACKGROUND. Fanconi anaemia (FA) is an inherited form of aplastic anaemia.
OBJECTIVE. To document the demographics, presenting features, clinical signs and laboratory results of a cohort of South African FA patients.
METHODS. This was a retrospective, file-based study of patients (N=144) with a presumed or proven diagnosis of FA over 43 years. Results of laboratory investigations, including diepoxybutane (DEB) chromosome breakage and molecular studies, were analysed. Patients' physical features, presenting symptoms, outcomes and, where indicated, cause of death were evaluated.
RESULTS. More than 70% of the patients were of non-Caucasian decent (n=104). The median age at diagnosis was 82 months. The median survival post diagnosis was 30 months. Among the Caucasian patients, mean post-diagnosis survival was 98 months, opposed to 19 months in the non-Caucasian group. Approximately 60% of cases had a fatal outcome; >20% of cases (mostly non-Caucasian) were lost to follow-up. There was an equal male : female ratio. The presenting complaints related to bone marrow problems in >60% of the patients. Anthropometry revealed that >70% of patients were below the 10th percentile for height, weight and head circumference. The incidence of radial ray abnormalities, typical facies, small eyes or pigmentation abnormalities was >80%. A positive DEB test and hypoplastic bone marrow were found in >90% of patients. Death was commonly related to anaemia, bleeding and sepsis.
CONCLUSION. The patients in this cohort were similar to those described previously, although certain variations, such as a high incidence of intracranial bleeds and lower incidence of renal anomalies, were noted.
Fanconi anaemia (FA) is a rare, inherited, aplastic anaemia resulting from impaired DNA repair mechanisms that cause increased chromosome breakages, either spontaneous or induced.[1] The disease is genetically and phenotypically heterogeneous owing to mutations associated with at least 22 different genes.[1-4] FA may have a higher incidence in specific racial and ethnic groups, such as among people of Ashkenazi Jewish descent or Afrikaner ancestry[1] and in black populations in southern Africa,[3] possibly with different genes involved in each of these groups.[3] The disease is usually inherited in an autosomal-recessive manner, but an X-linked form has also been described.[4]
FA is characterised by various congenital abnormalities, which usually involve growth abnormalities, skin pigmentation and anomalies of the skeletal, cardiac and renal systems; in some cases, the gastrointestinal and central nervous systems are also affected.[1,4-7] Aplastic anaemia or bone marrow failure is a hallmark of the disease, with pancytopenia, acute myeloid leukaemia and myelodysplastic syndromes being observed.[3,5,8] Clinical signs related to these blood diseases are often the presenting complaints and the diagnosis of FA is usually made only once the patient has been thoroughly evaluated; the minority of patients are diagnosed as a result of congenital abnormalities or an index case, such as an affected relative.
The aim of this study was to describe patients with FA assessed at the Universitas Academic Hospital in Bloemfontein, South Africa (SA) between 1974 and 2017. Their demographic information, presenting complaints, physical features, outcomes and the results of selected initial laboratory tests and special investigations are presented.
Methods
Data on patients from central SA with a presumed diagnosis of FA have been collected prospectively since the mid 1970s. Results from an earlier analysis[9] suggested the incidence of FA to be higher than normal in the area drained by the Universitas Academic Hospital (the Free State and Northern Cape provinces, certain areas of the North West and Eastern Cape provinces, and Lesotho).
Data were collected from all patients with a proven diagnosis of FA based on clinical signs, laboratory investigations and diepoxybutane (DEB) or molecular studies, as part of their routine clinical care. Patients who had classic clinical signs associated with bone marrow failure but for whom DEB or molecular studies were negative, had not been performed or had failed, were also included in the analysis. All patients were seen by the Paediatric Haematology and Oncology service, where the patient files were retained.
In the past, diagnosis was based on characteristic clinical findings and DEB testing was a possible confirmatory test. Today, the diagnosis rests on a high index of suspicion in patients who present with aplastic anaemia and classic anomalies. Molecular analysis is available for the most commonly identified mutations in certain population groups, but as comprehensive molecular analysis is not yet available diagnostically, such an analysis is used to confirm the diagnosis. Siblings of patients are actively tested if there is any possibility that they may be affected.
In the early years, treatment was purely supportive and included blood products and anabolic steroids. Later bone marrow transplants became an option to manage the bone marrow failure element of FA. Currently our indigent patients are offered improved supportive care and for patients with a matched sibling donor, bone marrow transplant is an option. In funded patients, an unrelated bone marrow transplant is possible, depending on the availability of finances; however, many patients do not progress to a transplant.
Ethical considerations
Approval to conduct the study was obtained from the Ethics Committee of the Faculty of Health Sciences, University of the Free State (ref. no. ETVOS 158/08).
Results
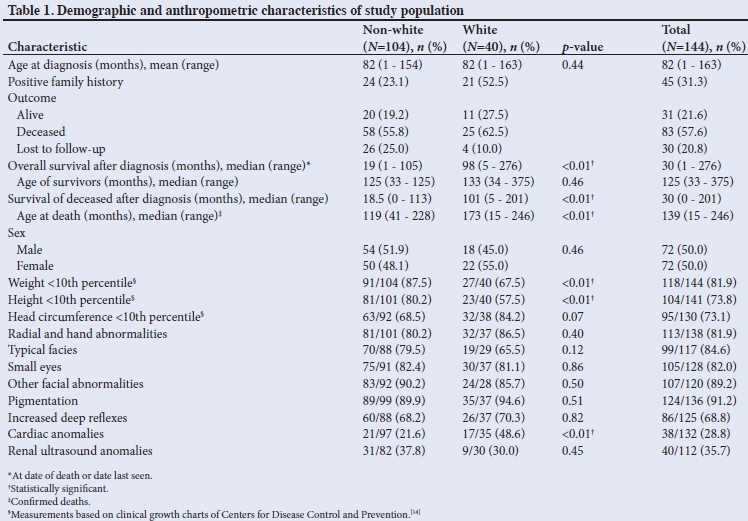
A total of 157 patients were diagnosed with FA during the 43-year period for which data were available. Only rudimentary notes were available in 13 cases, which were subsequently excluded from the analysis. Of the remaining 144 patients, 40 were white and 104 non-Cwhite (Table 1). White refers to individuals who are of European descent; non-white refers to individuals from indigenous black populations of southern Africa or of mixed ancestry.
Demographic and anthropometric characteristics
The mean age at diagnosis was 82 months (range 1 - 163 months), with no difference between the two study groupings. Female patients were generally older than male patients at presentation. Approximately a third (31.3%) of the patients had close relatives affected by the disease (male 21/144; female 24/144); more patients with affected relatives appeared to have been diagnosed during the earlier years of the data collection period. A positive family history appeared to be twice as common among white patients compared with the non-white group (52.5% v. 23.1%; p<0.01).
Male and female patients were equally distributed in the total study population, although the white group included slightly more female than male patients (55.0%). Approximately a third of the non-white patients were from Lesotho, whereas another third came from the QwaQwa region of the Free State province (bordering Lesotho and KwaZulu-Natal). The remainder were from our extended drainage area. The Caucasian patients did not have a geographic predilection, although the majority were of Afrikaner ancestry.
The median overall survival post diagnosis (until date of death or date last seen) was 30 months. Median survival among the white patients was 98 months compared with 19 months among non-white patients (p<0.01). The median age at death for the study population as a whole was 139 months; the median age at death among white patients was 173 months compared with 119 months among the non-white group (p<0.01).
As shown in Table 1, more than 70% of the study population were below the 10th percentile for weight, height and head circumference. With regard to weight, 87.5% of the non-white and 67.5% of the white patients were below the 10th percentile (p<0.01). The difference in height between the two groups was also statistically significant. Female patients were more likely to have an abnormal head circumference (<2nd percentile), and indeed all the white female patients presented with small heads. For approximately half of the patients all three anthropometric measurements were below the 10th percentile; this observation was more prevalent among non-white and male patients.
As of August 2017, 21.6% of the patients were confirmed to be alive and 57.6% had died. Approximately a fifth (20.8%) of the patients had been lost to follow-up and therefore their outcomes were unknown. Of this group, the majority were from the non-white cohort.
Thumb (radial) and hand abnormalities
A total of 113 (81.9%) patients had documented abnormal thumbs or hands. Only 15% of the white patients had normal thumbs, compared with 20% of the non-white group.
Facial abnormalities
The characteristic facies associated with FA (small eyes, low hairline, triangular face and puckered mouth) were observed in 84.6% of patients, with a higher percentage among non-Cwhite patients. Small eyes or apparent microphthalmia was seen in >80% of the patients, with a similar distribution between the two groups. Other facial abnormalities, such as hypertelorism, hypotelorism, strabismus, epicanthic folds, ptosis and a flat, wide nasal bridge, occurred in >85% of the patients. These findings were subjective and not measured according to standard dysmorphic guidelines.
Skin pigmentation
More than 90% of the patients had some form ofpigmentation abnormality, with >50% of these patients showing both hyper- and hypopigmented skin lesions. Only 30% had either hyper- or hypopigmented lesions, with hypopigmentation alone being uncommon (<1%). No substantial difference was observed in the pigmentation pattern across the two groups, although an absence of pigmentation abnormality was more common in non-white patients (10% v. 5%).
Abnormal tendon reflexes
Approximately 70% of all the patients had brisk reflexes. Hyperreflexia occurred in 68.2% and 70.3% of the non-white and white groups, respectively.
Congenital cardiac abnormalities
A cardiac lesion was found either clinically or on transthoracic echocardiography in 38 patients (28.8%). Table 1 shows that 21.6% of non-white and 48.6% of white patients presented with cardiac anomalies (p<0.01).
Renal anomalies
Renal abnormalities were found in 40 of 112 patients (35.7%) based on renal ultrasound investigation. A slightly higher incidence was seen among non-white patients (37.8% v. 30.0%).
Causes of death
As shown in Table 2, the most common causes of death were related to bleeding, anaemia and sepsis. In the cases in which bleeding occurred (n=30), 13 were due to proven or presumed intracranial bleeding, with a similar incidence across the two racial groups. Sepsis as the primary cause was the second most common cause of death (16.9%). Malignancy as cause of death occurred significantly more often in white (40.0%) than in non-white patients (6.9%).
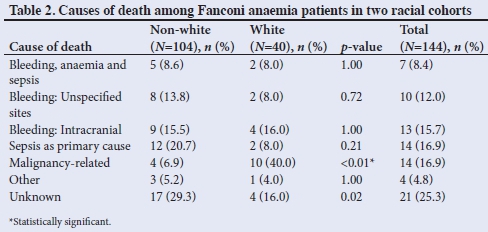
Cause of death was undetermined in 29.3% of cases in the non-white group, compared with 16.0% in the white group.
Presenting signs and laboratory tests
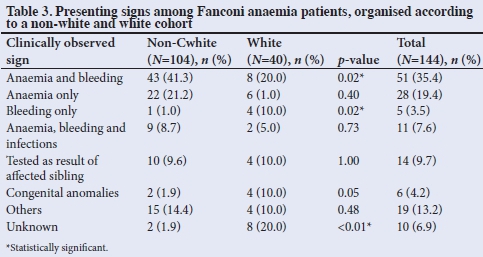
FA is associated with varied presenting signs. Table 3 shows that in this study population, 72.1% (75/104) ofthe non-white patients presented with signs related to bone marrow problems (anaemia, bleeding and infections) compared with only 50.0% (20/40) of the white patients. From the total study population, only 14 patients (9.7%) were diagnosed after a sibling had presented with FA. Congenital abnormalities were the primary reason for evaluation in 6 patients (4.2%). A statistically significant difference between the two groups was noted in those who presented with anaemia and bleeding (p=0.02) compared with bleeding only (p=0.02).
The results of the initial laboratory investigations are shown in Table 4. A haemoglobin level <10 g/dL at presentation was seen in 78.0% of the patients. In the white group, slightly more than 50% of the patients had a low haemoglobin level on presentation, opposed to 88.1% of the non-white group (p<0.01). Haemoglobin levels <7 g/dL occurred in 55% of the patients, with the incidence three times higher in the non-white group compared with the white group. Low haemoglobin levels were more common among female patients.
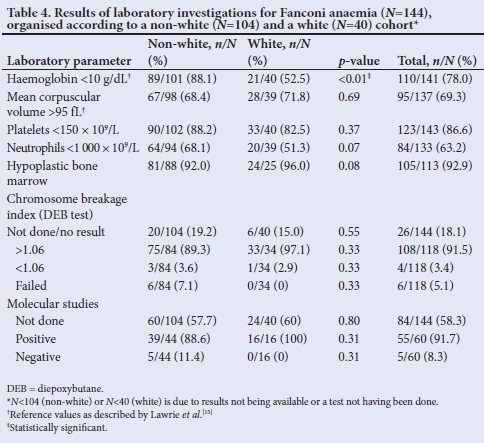
Full blood counts (FBC) often revealed macrocytosis and the mean corpuscular volume (MCV) was elevated in 69.3% of the patients (>95 fL). The occurrence of an increased MCV was similar in the two groups. The platelet count was below normal (150 χ 109/L) in 86.6% of the patients, with a similar incidence across the two racial groups. A platelet count of <50 χ 109/L was seen in 70% of patients, with the incidence higher among non-white patients (~80%) than among the white group (<50%). The white cell count (WCC) is often affected in FA and the neutrophil count was <1 000 χ 109/L in 63.2% of patients. More non-white patients presented with neutropenia than white patients (68.1% v. 51.3%). For the total group, more than 50% of patients presented with below-normal findings for all three haematological parameters (haemoglobin, platelet counts and neutrophil count), with a higher incidence in female and non-white patients.
A bone marrow aspiration was performed in 113 patients, with hypocellular samples found in 105 (92.9%). The remaining patients presented with either normal or hypercellular bone marrow. Approximately 85% of the non-white patients had a bone marrow aspiration performed at presentation, compared with less than 65% among the white group.
A DEB test had been performed in more than 80% (n=118) of the total study population, with a chromosome breakage index of >1.06 (positive DEB test) seen in 91.5% of these patients. Positive DEB results were obtained in 97.1% of Caucasian and 89.3% of non-white patients.
Molecular studies for FA are specific and the abnormalities in the SA population are generally seen in two genes, namely FANCG in black patients (c.637_643delTACCGCC) and FANCA in individuals of Afrikaner ancestry (deletion of exons 12-31, 11-17, 13-32, 12-18, 12-17, 11-12 and c.3398delA.
These mutations are especially common in the latter group and are due to a founder mutation. Molecular studies were performed in only 60 patients (41.7%). Of these, 55 (91.7%) tested positive for the relevant mutations. Isolated molecular studies were performed in the earlier years of the data collection period, but became routine only from about the middle of 2010. There are still cases for whom molecular testing is not performed, which may be due to inappropriate laboratory handling of samples or patients passing away before the studies can be performed.
A number of patients were not homozygous for the common deletions and presented with other associated molecular abnormalities. Six of the non-white patients were heterozygous for the 637_643 deletion in FANCG, whereas among the white patients, two common deletions were detected (exons 12-31 and 11-17). Approximately 90% of the non-white patients were homozygous for the FANCG 637_643 deletion. In the white group, mutations were identified in 31 of 32 alleles, with 7 patients being homozygous and eight heterozygous.
Discussion
Knowledge of FA has expanded substantially since the 1980s and many aspects of the diagnosis and management of the disease have changed. Today, the gold standard for diagnosis is molecular DNA analysis following presentation of typical physical features, whereas diagnosis was previously based only on physical features, with or without the DEB test and typical haematological features used for confirmation.
The median age at diagnosis of the patients in this study was 82 months (6.8 years), which was younger,[1,4,5] similar to[6,10] and older[8] than in comparable studies. The distribution of male and female patients was equal and similar to previous SA studies.[10,11] Despite the expected equal distribution, Shimamura and Alter[6] reported a significant increase (p<0.001) in the male:female ratio (1.2 : 1) among their FA patients, but could not provide an explanation for this finding.
Slightly more white patients had a fatal outcome, although a larger percentage of the non-white patients had been lost to follow-up, which could have affected the data. Figures from other SA studies reported death rates ranging from 68% to 81%,[8,10] compared with our rate of 57%. Many patients died at home, and consequently the cause of death could not be confirmed. Given the results in Table 2, it is presumed that most of these deaths were related to infections or bleeding. More than a third of the deaths were related to haemorrhage, with proven or presumed intracranial bleeding accounting for more than 40% ofthese (15.7% of all deaths). These figures are lower than those previously reported.[8,10] Malignancy-related deaths were ~six times more common in the white group, with leukaemia being four times more common in this group than among black patients.
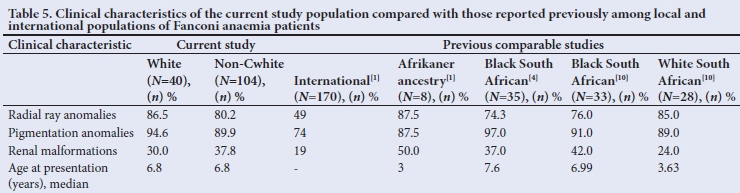
Physical abnormalities are common in children with FA and numerous abnormalities, varying in degree from obvious to subtle, have been reported.[1,4,6-8,10] The major abnormalities, with results for this cohort shown in Table 1 and compared with previously reported cohorts in Table 5, are discussed in the subsequent sections.
Thumb, radial and hand abnormalities
Radial abnormalities include absent radii and thumbs, duplicated thumbs, abnormal thumbs and hypoplastic thenar eminences. The thumbs may also be digitalised. Further radial abnormalities include absent or abnormal radial pulses. The ulnar side of the hand may also show hypothenar wasting.[4,6,7] Rosendorff et al.[11]reported radial abnormalities in 67% of their study population (of Afrikaner ancestry), compared with an incidence of 86.5% seen in our cohort. Similar to previous reports,[3,8,10] hand abnormalities occurred in 80.2% of the non-white patients. It is important to keep in mind that these signs may be subtle and should be actively sought.
Facial abnormalities
In addition to the typical facies of FA, a number of abnormalities of the eye are seen in FA, including epicanthic folds, up-slanting or short palpebral fissures and ptosis.[4,7,10] Similar to findings reported previously,[8,10] microphthalmia was noted in more than 80% of our patients, with little difference between the two racial groupings. In contrast, a substantially lower incidence of eye abnormalities (~20%) has been reported internationally in FA patients.[6]
Pigmentation abnormalities
Hyper- or hypopigmented skin lesions are common in children with FA and vary widely in number and distribution.[1,4,6-8,10,11] More than 90% of the patients in this study had some form of pigmentary changes, which is within a similar range as reported by other studies (71 - 100%).[4,8,10,11] Pigmentation abnormalities are an important sign to look for in aplastic anaemia patients when FA is suspected.
Congenital cardiac abnormalities
Several cardiac abnormalities in FA have been described,[1,4,6-8] varying from absent radial pulses to complex cardiac lesions, although this category is not consistently mentioned in reports.[11] In the present study, 28.8% of patients presented with a cardiac lesion, which is notably higher than in previous reports.[6,8]
Abnormal deep reflexes
Many children with FA present with brisk reflexes that affect either multiple or single deep-tendon reflexes. In our cohort, brisk reflexes were seen in 68.8% of the patients. Central nervous system abnormalities and hyperreflexia have been reported in 29 - 61% of SA patients with FA.[8,10]
Renal anomalies
Typical renal abnormalities seen in FA include pelvic, horseshoe and hypoplastic kidneys, duplex collecting systems and ectopic kidneys,[1,4] with an incidence of 20 - 50%.[1,4,6,8,11] Abnormal findings on renal ultrasound were seen in 35.7% of our patients. The slightly higher incidence of renal abnormalities among non-Cwhite patients is similar to previous reports.[10] In a recent study, Feben et al.[1]found renal anomalies in 50% of their patients of Afrikaner ancestry (n=8), compared with 30.0% (n=40) in our study.
Cardiac and renal abnormalities with concomitant limb abnormalities, and in some cases tracheo-oesophageal anomalies, should direct one to consider VACTRL association (vertebral defects, anal atresia, cardiac defects, tracheo-oesophageal fistula, renal anomalies and limb abnormalities) in these patients. Similarly, in children diagnosed with VACTRL association, FA should be excluded with appropriate testing.
Haematological abnormalities
Patients with FA often present with pancytopenia. Therefore, an FBC and a bone marrow aspiration and biopsy are part of the evaluation process.[1,4-8,10] Bone marrow studies may be omitted in very young patients who present with congenital anomalies, or those who have normal FBC parameters but are being investigated because of affected close relatives or siblings. This may explain the lower incidence of bone marrow examinations performed among white patients in our analysis.
The incidence of haemoglobin values <10 g/dL at presentation (78.0%) was higher than reported by Rosendorff et al.[11](67%), although a similar incidence (91 - 100%) has been reported previously.[10] The incidence of low haemoglobin levels among white patients and also the number of patients with haemoglobin levels <7 g/dL were lower than previously reported.[10]
Macrocytosis seen in an initial FBC may suggest FA. MCV was raised in 69.3%% of all patients in our study. Macrocytosis and elevated MCV values have been mentioned in relation to FA before, although no actual incidence has been reported.[1,5] Macdougall et al.[8,10]found an incidence of elevated MCV values ranging from 79% to 100%. The lower incidence observed in our study might be attributed to some patients having been referred because of family histories or congenital abnormalities and diagnosed while asymptomatic and in the pre-anaemic phase.
The low platelet count in 86.6% of children at presentation was similar to previously reported results.[8,10,11] However, at a cut-off level of 50 χ 109/L, 68% of the patients presented with low platelet counts, which is a lower incidence than reported previously when this reference point is used.[10]
Low WCCs have been reported in 72 - 100% of patients with FA.[10,11] Our finding regarding neutropenia (63.2%) is similar to what has been reported by Macdougall et al.[8]in non-white patients (60%).
Results from bone marrow investigations were similar to findings reported by Feben et al.[5]Other authors referred to bone marrow studies, but the incidence of hypoplasia was not evaluated further.[8,10] The DEB test was long regarded as yielding definitive diagnostic results, but problems in performing the test have since been identified. In some patients, DEB was not requested as mutation determination was undertaken to confirm the diagnosis. We found a high incidence of positive DEB results (97.1% in white and 89.3% in non-white patients). The rate in the white patients is similar to previous reports.[9,12] In non-white patients, positive DEB results have been reported in up to 83% of cases,[8,10] which is lower than our rate. Not all patients for whom FA is suspected based on clinical and haematological signs, will have a positive DEB test, and it is therefore important to keep in mind that negative or non-significant DEB results do not necessarily exclude a diagnosis of FA.[13] Laboratory personnel indicate that the most common reason for failure of the DEB test is the absence of mitotic activity because of a low WCC.
Molecular abnormalities
Molecular (DNA-based) studies for FA are very specific and many complementation groups have been described,[1-7] with the recent literature asserting that mutations occur in at least 22 different FA genes. In the SA population, two genes are commonly involved in FA and a number of mutations in either can result in FA. The common mutations identified are in the FANCA gene in patients of Afrikaner ancestry and in the FANCG gene in patients from black populations. In SA white patients, the common deletions involve exons 12-31, 13-32, 11-17 and 12-17, and also c.3398delA.[1] In the non-white (black) patients in our study, 75.0% were homozygous for deletion of c.637_643delTACCGCC, similar to findings reported by Feben et al.[1]All of the white patients tested for a genetic abnormality presented with deletions in the FANCA gene, although at various loci. Our study showed the proportion of common deletions (del13-31, 11-17 and c.3398delA) to be ~10% lower than what has been reported previously.[1]
Malignancies and other abnormalities
Among the cohort of FA patients in our study, 16 (11.1%) either had myeloid leukaemia at presentation or developed it at some stage during their follow-up. Not all the leukaemias were further delineated, as blast counts were often low. The incidence of leukaemia in patients with FA varies from 0% to 14%.[5-7,10] Other malignancies were documented in four patients (three cases of oesophageal carcinoma and one of vulvar carcinoma), which was very low compared with what has been described in literature.[6,7] However, the finding may be related to patients being lost to follow-up, dying at home due to unknown causes, or the mortality rate being so high that patients died before they developed malignancies.
Other congenital abnormalities included congenital hip dislocation («=2), tracheo-oesophageal fistula («=3) and single cases of pyloric stenosis, scimitar syndrome and an unexplained laparotomy as a neonate. Deafness was uncommon in this cohort and occurred in only one patient, which was notably lower than what has been previously reported.[1,3] It is possible that not all patients with hearing loss were identified, as the facilities for auditory examination are limited and we therefore test only if deafness is a presenting complaint.
Only a small number of patients (n=14) were identified after a sibling presented or was diagnosed with FA. This appears to be a missed opportunity for early diagnosis.
Clinical profile of a typical Fanconi anaemia patient in South Africa
The following characteristics are typically seen in local FA patients:
• Between 6 and 7 years of age
• Family history of FA likely in Caucasian patients
• Present with anaemia, bleeding or sepsis
• Underweight, short stature and small head
• Hand abnormalities, usually involving the thumbs or radial aspect of the hands
• Small eyes and other facial abnormalities
• Hyper- and hypopigmented skin lesions
• Brisk reflexes or an increased incidence of deep-tendon reflexes
• Likelihood of abnormal renal ultrasound or cardiac abnormality >25%
• Pancytopenia with a low haemoglobin level, a high MCV, and low platelet and neutrophil counts at presentation
• Hypoplastic bone marrow
• A positive chromosomal breakage index (>1.06) as determined with the DEB test, irrespective of race
• Non-Caucasian (black) patients likely to be homozygous for 637_643del in FANCG; varying genetic results in patients of Afrikaner ancestry, although many are homozygous for deletion exons 12-31, 11-17 or c.3398delA in the FANCA gene
• Death expected to occur by 12 years of age, usually owing to a combination of bleeding (most likely an intracranial incident), anaemia and sepsis.
Study limitations
Owing to the retrospective nature of this analysis, certain limitations were encountered. Some patients' files had missing clinical information or data that could not be verified. Regardless, this is the largest series of FA patients reported from a single unit in SA.
Conclusion
Overall, our findings were similar to what has been reported in other local and international studies, also with regard to the results of special investigations. The high incidence of the 637_643del mutation in the FANCG gene among non-white (black) patients in our study may be a valuable diagnostic test in this group of patients. Various deletions in the FANCA gene were seen in patients of Afrikaner ancestry, which may present more challenging diagnostic results. Standard diagnostic tests may detect FANCA deletions, but the interpretation of the compound heterozygotes is complex and needs to be performed by a laboratory with suitable expertise and ideally under the guidance of a geneticist.
FA is a rare disease, which is difficult to diagnose without clinical suspicion. It should be a differential diagnosis in all patients with aplastic anaemia, especially among those with congenital abnormalities of the face, hands, thumbs and associated cardiac or renal anomalies. Infants with suspected VACTRL anomalies should also be considered for FA. In both groups, the appropriate investigations need to be performed, supplemented with a genetic consultation and evaluation if necessary. Treatment is preferably by a paediatric haematologist or oncologist.
Acknowledgements. The author acknowledges the contribution of the following people with regard to data collection over the past 43 years: Drs S Smith, C Jordaan, C Havenga, C Weyers, J du Plessis, K Rautenbach, N McGill, R Berry, S Stannard, E Top, C van Niekerk, Sister L van Niekerk, and all the other personnel in the Paediatric Haematology and Oncology ward and Outpatients unit at the Universitas Academic Hospital. Dr B Henderson is thanked for assistance with the genetic aspects and editing of the article. The genetic laboratories at Universitas Academic Hospital and the National Health Laboratory Service (Johannesburg) are thanked for analysis of DEB tests and molecular studies. Dr D Struwig (Faculty of Health Sciences, University of the Free State) is thanked for technical and editorial preparation of the manuscript and the assistance of Prof. G Joubert (Department of Biostatistics, University of the Free State) in statistical analysis is acknowledged.
Author contributions. Sole author.
Funding. None.
Conflicts of interest. None.
References
1. Feben C, Haw T, Stones D, et al. Fanconi anaemia in South African patients with Afrikaner ancestry. S Afr J Child Health 2017;11(3):141-145. https://doi.org/10.7196/SAJCH.2017.v11i3.1312 [ Links ]
2. Li N, Ding L, Li B, Wang J, DAndrea AD, Chen J. Functional analysis of Fanconi anemia mutations in China. Exp Hematol 2018;66:32-41. https://doi.org/10.1016/j.exphem.2018.07.003 [ Links ]
3. Morgan NV, Essop F, Demuth I, et al. A common Fanconi anemia mutation in black populations of sub-Saharan Africa. Blood 2005;105(9):3542-3544. https://doi.org/10.1182/blood-2004-10-3968 [ Links ]
4. Feben C, Kromberg J, Wainwright R, et al. Phenotypic consequences in black South African Fanconi anemia patients homozygous for a founder mutation. Genet Med 2014;16(5):400-406. https://doi.org/10.1038/gim.2013.159 [ Links ]
5. Feben C, Kromberg J, Wainwright R, et al. Hematological consequences of a FANCG founder mutation in Black South African patients with Fanconi anemia. Blood Cells Mol Dis 2015;54(3):270-274. https://doi.org/10.1016/j.bcmd.2014.11.011 [ Links ]
6. Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev 2010;24(3):101-122. https://doi.org/10.1016/j.blre.2010.03.002 [ Links ]
7. Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res 2009;668(1-2): 4-10. https://doi.org/10.1016/j.mrfmmm.2009.01.013 [ Links ]
8. Macdougall LG, Greeff M, Rosendorff J, Bernstein R. Fanconi anemia in black African children. Am J Med Gen 1990;36(4):408-413. https://doi.org/10.1002/ajmg.1320360408 [ Links ]
9. Marx MP, Smith S, Heyns AD, Van Tonder IZ. Fanconi's anemia: A cytogenetic study on lymphocyte and bone marrow cultures utilizing 1,2:3,4-diepoxybutane. Cancer Genet Cytogenet 1983;9(1):51-59. https://doi.org/10.1016/0165-4608(83)90024-9 [ Links ]
10. Macdougall LG, Rosendorff J, Poole JE, Cohn RJ, McElligott SE. Comparative study of Fanconi anemia in children of different ethnic origin in South Africa. Am J Med Gen 1994;52(3):279-284. https://doi.org/10.1002/ajmg.1320520306 [ Links ]
11. Rosendorff J, Bernstein R, Macdougall L, Jenkins T. Fanconi anemia: Another disease of unusually high prevalence in the Afrikaans population of South Africa. Am J Med Gen 1987;27(4):793-797. https://doi.org/10.1002/ajmg.1320270408 [ Links ]
12. Auerbach AD, Rogatko A, Schroeder-Kurth TM. International Fanconi Anemia Registry: Relation of clinical symptoms to diepoxybutane sensitivity. Blood 1989;73(2):391-396. https://scinapse.io/papers/1562232013 [ Links ]
13. Rosendorff J, Bernstein R. Fanconi's anemia - chromosome breakage studies in homozygotes and heterozygotes. Cancer Genet Cytogenet 1988;33(2):175-183. https://doi.org/10.1016/0165-4608(88)90027-1 [ Links ]
14. Centers for Disease Control and Prevention. Clinical growth charts. https://www.cdc.gov/growthcharts/clinical_charts.htm (accessed 4 April 2018). [ Links ]
15. Lawrie D, Payne H, Niewoudt M, Glencross DK. Observed full blood count and lymphocyte subset values in a cohort of clinically healthy South African children from a semi-informal settlement in Cape Town. S Afr Med J 2015;105(7):589-595. https://doi.org/10.7196/SAMJnew.7914 [ Links ]
 Correspondence:
Correspondence:
D Stones
StonesDK@ufs.ac.za
Accepted 13 May 2019


{kind=link}
{kind=link}