










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Child Health
versão On-line ISSN 1999-7671
versão impressa ISSN 1994-3032
S. Afr. j. child health vol.13 no.4 Pretoria Dez. 2019
http://dx.doi.org/10.7196/sajch.2019.v13i4.1630
RESEARCH
The use of hydroxyurea in sickle cell disease: A single tertiary centre experience at the National Hospital, Abuja, Nigeria
O OniyangiI; A B OyesakinII; G O EzehII; E J OkonI; T T WakamaIII; J A F MomohIV; A B AkanoV; H A AikhionbareI
IMBBS, FWACP (Paeds); Department of Paediatrics, National Hospital, Abuja, Nigeria
IIMBBS, FMC (Paeds); Department of Paediatrics, National Hospital, Abuja, Nigeria
IIIMBBS, FMC (Path); Department of Haematology, National Hospital, Abuja, Nigeria
IVMBBS, FWACP (LabMed); Department of Chemical Pathology, National Hospital, Abuja, Nigeria
VMBBS, FMC (Radiol); Department of Radiology, National Hospital, Abuja, Nigeria
ABSTRACT
BACKGROUND. Hydroxyurea (HU) has been found to be beneficial in sickle cell disease (SCD), reducing the occurrence of severe manifestations of the disease such as painful crises and blood transfusions. Although a standard of care for SCD in the developed countries of the world, limited data are available on its use in Africa.
OBJECTIVES. To review indications of the use of HU, laboratory monitoring and outcome in children with SCD.
METHODS. A retrospective review of 74 patients treated with HU (15 - 30 mg/kg/day) for a minimum of 6 months. The main outcome measures were indications for use of HU, haemoglobin level (Hb), packed cell volume (PCV), white cell count (WBC), absolute neutrophil count (ANC), serum alanine aminotransferase (ALT) and creatinine. Descriptive statistics were expressed as means ±2 standard deviations. Data were compared pre and post HU therapy, using the chi-square and Student's t-test as appropriate.
RESULTS. The 74 patients constituted 7.26% of the SCD clinic population and were aged 2.25 - 16 years (mean (SD) 8.48 (3.67)) and were on HU therapy for 0.5 - 4.8 years (mean (SD) 1.72 (1.12)). The haemoglobin genotypes were Hb SS 72 (98.7%); Hb SC and Hb SS+F 1 (1.35%) each. Indications for HU use were abnormalities of transcranial Doppler ultrasound (TCD) 39 (52.7%), multiple vaso-occlusive crises (VOC) 18 (24.3%), strokes 14 (18.9%), as well as repeated blood transfusions 9 (12.2%) and hospital admissions 7 (9.5%). Some patients had more than one indication. There were significantly fewer TCD abnormalities, VOC, strokes, splenic sequestrations, blood transfusions and hospital admissions following use of HU. The mean Hb and PCV increased while WBC and ANC decreased. Occurrence of acute chest syndrome, priapism, serum creatinine, ALT and platelet levels were not affected.
CONCLUSIONS. Hydroxyurea therapy reduces the occurrence of severe manifestations and improves laboratory parameters in children with SCD.
The drug hydroxyurea (HU) has been found to be beneficial in sickle cell disease (SCD).[1-3] Systematic reviews of the effectiveness of HU have shown the drug to reduce the rate of painful crises, acute chest syndrome, number and duration ofhospital attendances and admissions; blood transfusions and mortality in SCD; as well as reducing abnormal transcranial Doppler ultrasound (TCD) values towards normal, thus decreasing the risk of stroke.[2,3] In addition, trials have shown it is well tolerated, does not impair growth or development in children, and can safely be used from the age of 9 - 18 months.[4]
There is limited use of HU for SCD in Africa[5] although there are now a number of ongoing clinical trials investigating its effectiveness and safety.[6,7] Furthermore, the care guidelines for some African countries including Nigeria recommend the use of HU for certain clinical conditions.[8] Despite long-term use of the drug for SCD in more developed parts of the world,[11]its use is not yet a standard of care in Africa,[5] where the burden of SCD is high, owing to lack of, or inadequate availability, cost and access to the drug, as well as physician/health facility capacity to manage and monitor people on the medication.[5] In Nigeria, the use of HU for SCD is low.[9,10]
The present report on the use of HU in children with SCD is intended to raise awareness of the potential use of the medication. The objectives are to discuss patient selection, laboratory monitoring and the outcome of children with SCD after a minimum of 6 months of HU therapy.
Methods
This is a retrospective review of children aged 2 - 16 years with SCD attending the paediatric sickle cell clinic of the National Hospital, Abuja, Nigeria, over a 4-year period from January 2010 - January 2014. Their haemoglobin genotype was determined by cellulose acetate haemoglobin electrophoresis.
The routine protocol of care for children with SCD in this hospital includes a 2 - 3-monthly clinical review in the outpatient clinic, counselling, immunisations, monitoring of laboratory parameters (full blood count (FBC), serum alanine aminotransferase (ALT) and creatinine), folic acid supplementation, administration of proguanil anti-malarial (Nigeria is holoendemic for malaria) and penicillin prophylaxis. In addition, children with the following severe manifestations of SCD were placed on HU therapy:
• 3 or more episodes of severe vaso-occlusive crises (VOC), hyperhaemolytic crises, blood transfusions or hospital admissions in the preceding 12 months
• at least 1 episode of stroke, acute chest syndrome (ACS), sequestration crises or priapism
• abnormalities of transcranial Doppler ultrasound studies (TCD) (cm/sec.) -high >200, conditional (170 - 199) and discrepant (>50% difference between both sides).
Parents, caregivers and children as appropriate were counselled about the use of HU. The medication was started at 15 mg/kg as a daily single dose, increasing to a maximum of 30 mg/kg. Increments in drug dosage were done by 5 - 10 mg/kg every 2 months until the maximum tolerated dose was achieved. Follow-up was after 2 weeks, then monthly for 2 months and then every 3 months. A full clinical evaluation was performed at each visit, and specific questions relating to the potential side-effects of HU were asked. Haemoglobin (Hb), packed cell volume (PCV), white cell count (WBC), (absolute neutrophil count) ANC, platelet count, serum alanine aminotransferase (ALT) and creatinine were done before commencement of HU and at each follow-up visit or as required by the physician. The HU dosages were approximated to the available capsule sizes of 100 mg, 250 mg and 500 mg, and compounded as needed. All blood samplings and TCD evaluations were done when the children were in steady state.[8] A repeat evaluation of the indication for HU was done after 6 months.
During the period, 74 children were treated with HU. In the same period, a total of 537 children were seen at the SCD clinic of the hospital. Hospital records of the 74 patients were reviewed.The results were presented as simple frequencies and percentages and shown in tables. Descriptive statistics were expressed as means ± 2 standard deviations. Associations between variables were done by the chi-square test and Student's i-test as appropriate. A p-value <0.05 was considered statistically significant.
Results
There were 42 boys and 32 girls (M:F = 1:1.3) aged 2.25 - 16 years, (mean (SD) 8.48 (3.67) years) and constituted 7.26% of the children seen at the SCD clinic. The haemoglobin genotypes were Hb SS 72 (97.3%), and Hb SC and HbSS + F 1 each (1.35%). The duration of use of HU was 0.5 - 4.8 years (mean (SD) 1.72 (1.12) years). Indications for HU use were abnormalities of TCD studies 39 (52.7%), multiple vaso-occlusive crises 18 (24.3%), strokes 14 (18.9%) and repeated blood transfusions 9 (12.2%) (Table 1). Some children had more than one indication, as shown in Table 1.
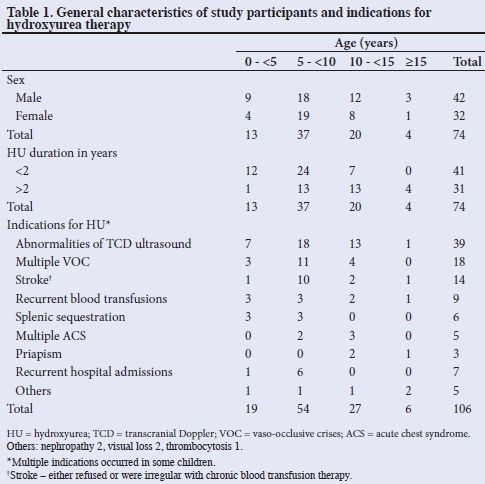
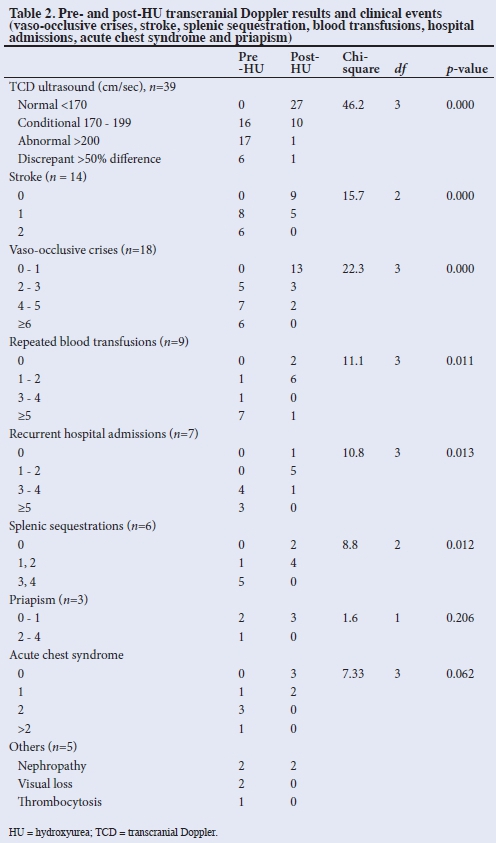
There were significantly fewer children (p<0.005) with abnormalities on TCD, as well as decreased number of episodes of the following clinical events post HU therapy: VOC, strokes, splenic sequestrations, blood transfusions and hospital admissions. There was no difference in the occurrence of ACS and priapism post HU therapy (Table 2).
The pre- and post-HU haemoglobin (Hb) (g/dL) concentrations were 5 - 10.7 (mean (SD) 7.64 (1.13)) and 5.7 - 11.9 (mean (SD) 8.66 (1.24)) respectively, (p=0.000). PCV was 16 - 30% (mean (SD) 23.01 (3.14)%) pre HU and 18.7 - 34% (mean (SD) 26.28 (3.53)%) post HU (p=0.000), indicating significant increases in the mean Hb and PCV levels post HU therapy (p<0.05). One child had a decrease in Hb and PCV levels and 5 children had no change in these parameters.
Pre- and post-HU WBC (x105/L) were 3 - 28.7 (mean (SD) 13.01 (6.05)); and 2.4 - 19.5 (mean (SD) 9.829 (3.75)), respectively (p=0.000). Pre- and post-HU ANC were 622 - 14 673 (mean (SD) 5 732.84 (2 948.61)) and 506 - 10 974 (mean (SD) 4 334.08 (2 111.95)) respectively, (p=0.0363), indicating that the mean post-HU WBC and ANC were significantly lower than the pre-HU levels.
Pre- and post-HU platelet counts (105/L) were 43.1 - 820 (mean (SD) 333.02 (169.6) and 97 - 659 (mean (SD) 289.24 (124.4)) respectively, (p=0.084). Four children had a decreased platelet count (x 105/L) to <150, and 2 with platelet counts <100 post HU therapy. These children were asymptomatic. Pre- and post-HU serum creatinine (mmol/L) were 7 - 97 (mean (SD) 32.07 (13.32)) and 15 - 167 (mean (SD) 38.77 (22.45)) respectively, (p=0.037). However, the mean serum creatinine levels were higher post HU but remained within the normal range of values. One child with markedly elevated[18] serum creatinine levels (167 mmol/L) post HU therapy had been previously diagnosed with sickle cell nephropathy. Serum ALT (mmol/L) pre and post HU were 2 - 89 (mean (SD) 20.04 (15.41)) and 4 - 372 (mean (SD) 30.25 (53.24)) respectively, (p=0.143). Two children had markedly elevated[18] ALT levels post HU.
Discussion
The use of HU, though a standard of care in high-income countries, is not routine in less-developed parts of the world.[5] In the present report, only a small proportion of children with SCD attending the outpatient clinic received HU; this may be due to physician/caregiver inexperience regarding patient selection for the use of HU. Furthermore, HU was not readily available or accessible at the time and was expensive. This scenario is similar to previous reports from sub-Saharan Africa,[5] including Nigeria,[9,10] where HU is seldom used for several reasons such as lack of (or inadequate) availability, the cost and access to the drug, and physician/health facility capacity. It is hoped that with increasing information and availability, the drug will be considered for use in SCD in the region. The duration of follow-up varied between 0.5 and 4.8 years (mean 1.72 years). It may be that this time period was insufficient to ascertain the efficacy of HU, especially as it relates to clinical events. However, a minimum period of 6 months is expected to show the effects of HU.[1,3]
Indications for HU use were mainly severe SCD complications based on available evidence.[2,3] In addition, limited physician and parent/caregiver capacity and experience in the use of the drug might have contributed to this patient selection as it was easier for both parents and physicians to agree on the use of the medication after a complication had occurred. This scenario is similar to other reports from Nigeria;[11-13] however, it may be a limitation to the interpretation of the study results, as the efficacy of HU cannot be wholly established from such a small part of the clinic size. HU indications in the present report were a reflection of the complications of SCD encountered in the outpatient clinic but are similar to other reports.[1-3,5,8,11]
In the present report, HU therapy was shown to significantly decrease the rates of severe complications of SCD such as VOC, repeated blood transfusions, hospital admissions and sequestration crises (p<0.05), but not priapism or ACS. In addition, there was an improvement in TCD values towards normal, as well as a decrease in occurrence of stroke. HU has previously been shown to be beneficial in reducing these severe complications of SCD in both children and adults.[1-3,5,11-13] Our results are similar to previous reports in this regard.
HU has been shown to decrease the occurrence of ACS in SCD,[1,2] and is indeed suggested as an indication for use of the drug by the Nigerian national guidelines for control and care of SCD,[8] but the present report did not show such benefit. A recent report also did not support the use of HU for ACS.[14] Although HU has no role in the management of acute priapism, its use has been suggested to prevent recurrences of this clinical event.[15,16] One case study reported recovery of erectile function with HU therapy in an adolescent with sickle cell anaemia following a prolonged episode of priapism and subsequent severe erectile dysfunction, as well as prevention of priapism episodes.[15] Another report has indicated prevention of recurrence of stuttering priapism by HU.[16] The small number of patients with ACS and priapism in the present report makes it difficult to come to any definite conclusions regarding the role of HU in these complications.
Furthermore, the dose and duration of HU treatment might be a contributory factor, although all children had been on the drug for upwards of 6 months, and indeed both children with ACS for up to 3 years.
Identifying children with SCD at risk of stroke by TCD abnormalities enables primary prevention of the event.[2,3,13,14]
In the present report, HU has been shown to reverse conditional and high TCD values towards normal, thus decreasing the risk of stroke, which is similar to previous reports[2,3,13,14] and underscores the importance of routine screening with TCD in SCD.
Stroke is a devastating complication of SCD, and TCD abnormalities identify children at risk. Several reports have shown that HU reduces the risk of stroke in children with SCD by improving the TCD velocities.[2,3,13,14] In the present report, HU therapy is shown to reduce the occurrence of stroke in SCD. Reports from Nigeria[13] and Jamaica[17] have shown similar findings. However, 5 of the 14 children in our report had 1 recurrence of stroke despite HU therapy for up to 1 year, as mentioned in other reports.[1,13,17] As children with stroke are prone to reccurrences,[1] this factor may create bias in the results of HU use in these patients. However, some reports[13,14,17] have indicated that although HU may not be the treatment of choice for prevention of secondary stroke in SCD, it is preferable to supportive care alone, especially in sub-Saharan Africa, where chronic blood transfusion therapy might not be feasible. Four of 6 patients with splenic sequestration had 1 repeat episode while on HU. Sequestration crisis is not a frequently reported indication for HU therapy, and it has been suggested that the drug may not be beneficial in this regard.[17]
Monitoring the effects of HU has always been a concern for both the healthcare provider as well as caregivers of children with SCD. We were unable to monitor either Hb F levels or mean cell volume (MCV), which are measures of response to HU as well as compliance with therapy.[19] Our results showed there were improvements in Hb and PCV levels following use of HU, as in other reports.[14,19,20] The significant decrease (p<0.05) in mean WBC and ANC measures of drug toxicity[19] are similar to previous reports.[14,19] HU is reported to be potentially toxic to both the liver and kidneys.[19] However, at the doses used in SCD, such toxic effects are uncommon.[19] In the present report, there was no elevation of ALT with HU therapy, as in previous reports. Although there were no differences in serum creatinine levels pre and post HU therapy, the overall mean levels did increase; this was due to an extremely high value in one child with previously diagnosed sickle cell nephropathy on follow-up, and may not be a true reflection of the study population.
Based on the results from the present study, the following indications are proposed for use of HU in SCD in Africa: abnormalities of TCD studies, >3 vaso-occlusive crises, 3 hospital admissions or blood transfusions withiin 12 months, at least 1 sequestration crisis and at least 1 stroke episode.
Conclusion
Hydroxyurea therapy appears to be beneficial in children with SCD in the study setting, with improvements in clinical events and minimal side-effects from treatment. Drug compliance which is required for optimal effects of the drug was not assessed in the study. Healthcare providers should consider using this drug in children with SCD. Further studies on side-effects and long-term use of the drug are necessary.
Declaration. None.
Acknowledgements. We gratefully acknowledge Professor E A Ameh of the National Hospital, Abuja, for his critical review of the manuscript. We acknowledge the contributions by all staff of the sickle cell clinic in the care of affected children and their families. We thank the children and their families who used the drug HU.
Conflicts of interest. None.
Author contributions. All authors contributed to the study. OO, ABO, GOE, EJO and HAA participated in patient recruitment and follow-up at the sickle cell clinic. TTW, JAFM and ABA participated in laboratory and radiological follow-up of the children. OO and TTW contributed to the design of the research and writing of the manuscript. All authors participated in writing various parts of the paper. All authors reviewed and approved the final manuscript.
Funding. None.
References
1. The management of sickle cell disease. National Institutes of Health. National Heart, Lung, and Blood Institute Division of Blood Diseases and Resources 2014. NIH Publication No. 02-2117. https://www.nhlbi.nih.gov/files/docs/guidelines/scmngt.pdf (accessed 21 October 2018). [ Links ]
2. Nevitt SJ, Jones AP, Howard J. Hydroxyurea (hydroxycarbamide) for sickle cell disease (Review). Cochrane Database of Systematic Reviews 2017, Issue 4, Art. no.: CD002202. https://doi.org/10.1002/14651858.cd002202.pub2 [ Links ]
3. Strouse JJ, Lanzkron S, Beach MC, et al. Hydroxyurea for sickle cell disease: A systematic review for efficacy and toxicity in children. Pediatrics 2008;122(6):1332-1342. https://doi.org/10.1542/peds.2008-0441 [ Links ]
4. Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: A multicentre, randomised, controlled trial (BABY HUG). Lancet 2011;377(9778):1663-1672. https://doi.org/10.1016/j.yped.2011.09.024 [ Links ]
5. Mulumba LL, Wilson L. Sickle cell disease among children in Africa: An integrative literature review and global recommendations. Int J Afr Nurs Sci 2015;3:56-64. https://doi.org/10.1016/j.ijans.2015.08.002 [ Links ]
6. McGann PT, Tshilolo L, Santos B, et al. Hydroxyurea therapy for children with sickle cell anemia in sub-Saharan Africa: Rationale and design of the REACH Trial. Pediatr Blood Cancer 2016;63(1):98-104. https://doi.org/10.1002/pbc.25705 [ Links ]
7. Galadanci NA, Abdullahi SU, Tabari MA, et al. Primary stroke prevention in Nigerian children with sickle cell disease (SPIN): Challenges of conducting a feasibility trial. Pediatr Blood Cancer 2015;62:395-401. https://doi.org/10.1002/pbc.25289 [ Links ]
8. Nigeria: National Guideline for the Control and Management of Sickle Cell Disease. Federal Ministry of Health, Nigeria, 2014. http://health.gov.ng/doc/SCDGuideline.pdf (accessed 21 October 2018). [ Links ]
9. Aliyu ZY, Babadoko AA, Mamman AI. Hydroxyurea utilization in Nigeria: A lesson in public health. Blood 2007;110:80. http://www.bloodjournal.org/content/110/11/80 [ Links ]
10. Galadanci N, Wudil J, Balogun T, et al. Current sickle cell disease management practices in Nigeria. Int Health 2014;6(1):23-28. https://doi.org/10.1093/inthealth/iht022 [ Links ]
11. Adewoyin AS, Oghuvwu OS, Awodu OA. Hydroxyurea therapy in adult Nigerian sickle cell disease: A monocentric survey on pattern of use, clinical effects and patient's compliance. Afr Health Sci 2017;17(1):255-261. https://doi.org/10.4314/ahs.v17i1.31 [ Links ]
12. Lagunju I, Brown BJ, Sodeinde O. Hydroxyurea lowers transcranial Doppler flow velocities in children with sickle cell anaemia in a Nigerian cohort. Paediatr Blood Cancer 2015;62(9):1587-1591. https://doi.org/10.1002/pbc.25529 [ Links ]
13. Lagunju IA, Brown BJ, Sodeinde OO. Stroke recurrence in Nigerian children with sickle cell disease treated with hydroxyurea. Niger Postgrad Med J 2013;20(3):181-187. https://www.ncbi.nlm.nih.gov/pubmed/24287747 [ Links ]
14. DeBaun M, Galadanchi N. Sickle cell disease in sub-Saharan Africa. https://www.uptodate.com/contents/sickle-cell-disease-in-sub-saharan-africa [ Links ]
15. Anele UA, Mack AK, Resar LMS, Burnett AL. Hydroxyurea therapy for priapism prevention and erectile function recovery in sickle cell disease: A case report and review of the literature. Int Urol Nephrol 2014;46(9):1733-1736. https://doi.org/10.1007/s11255-014-0737-7 [ Links ]
16. Al Jam'a AH, Al Dabbous IA. Hydroxyurea in the treatment of sickle cell associated priapism. J Urol 1998;159(5):1642. https://doi.org/10.1007/s11255-014-0737-7 [ Links ]
17. Ali SB, Moosang M, King L, et al. Stroke recurrence in children with sickle cell disease treated with hydroxyurea following first clinical stroke. Am J Hematol 2011;86(10):846-850. https://doi.org/10.1002/ajh.22142 [ Links ]
18. World Health Organization. The World Health Report 2003 - WHO toxicity grading scale for determining the severity of adverse events. Geneva: WHO, 2003. http://www.icssc.org/documents/resources/aemanual2003appendicesfebruary_06_2003%20final.pdf (accessed 21 October 2018). [ Links ]
19. Ware RE. Optimizing hydroxyurea therapy for sickle cell anemia. Hematology Am Soc Hematol Educ Program 2015;436-443. https://doi.org/10.1182/asheducation-2015.1.43643 http://asheducationbook.hematologylibrary.org/content/2015/1/436.full [ Links ]
 Correspondence:
Correspondence:
O Oniyangi
seyioniyangi@gmail.com
Accepted 4 April 2019

