










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Child Health
On-line version ISSN 1999-7671
Print version ISSN 1994-3032
S. Afr. j. child health vol.13 n.3 Pretoria Sep. 2019
http://dx.doi.org/10.7196/SAJCH.2019.v13i3.1636
CASE REPORT
A case report of a patient with Schimke immuno-osseous dysplasia and co-morbid Moyamoya Syndrome
R GovenderI; E NaickerII; K PillayIII
IMB ChB, DCH, FCPaeds, Cert Paed Neurol, MPhil; Department of Paediatric Neurology, Department of Paediatrics and Child Health, Inkosi Albert Luthuli Central Hospital, Durban, South Africa
IIMB ChB, DCH, FCPaeds, Cert Paed Nephrol; Department of Paediatric Nephrology, Department of Paediatrics and Child Health, Inkosi Albert Luthuli Central Hospital, Durban, South Africa
IIIMB ChB, DCH, FCPaeds; Westville Hospital, Durban, South Africa
ABSTRACT
Schimke immune-osseous dysplasia (SIOD) is a rare autosomal recessive disorder presenting with dysmorphic features, skeletal dysplasia, steroid resistance nephrotic syndrome and cellular immune insufficiency. Central nervous system complications such as Moya-moya syndrome have been reported as a co-morbidity. We describe a case study of a South African child who was diagnosed at the age of 5 years. She initially presented with short stature secondary to the skeletal dysplasia. She subsequently developed Moyamoya syndrome and steroid-resistant nephrotic syndrome. Genetic studies confirmed the presence of the c.1439 C>T mutation in the SMARCAL1 gene, which has been described in patients with SIOD previously. She died at the age of 7 years from an inter-current pneumonia.
Schimke immuno-osseous dysplasia (SIOD) is a multi-system progressive disorder. It was first described as a spondyloepiphyseal dysplasia by Schimke et al.[1] in 1971. It is characterised by autosomal recessive inheritance, spondyloepiphyseal dysplasia causing growth retardation, defective cellular immunity with episodic lymphopenia, nephrotic syndrome and progressive nephropathy leading to renal failure, pigmentary skin changes and progressive cerebrovascular disease complicated by cerebral infarcts and Moyamoya syndrome.[2]The neurological complications have been reported but are not well defined.
Case
The index patient is a South African child of distantly consanguineous parents. There was an antenatal assessment of a single umbilical artery and intrauterine growth restriction. She was born preterm at a gestational age of 29 weeks and birthweight of 700 g. She required respiratory support for hyaline membrane disease at birth. She was the second child of healthy parents. An elder sibling demised at day 10 of life from an unknown aetiology. She has a healthy brother who is now 17 years old.
Clinically, her weight and height were both <-3SD. She had dysmorphic features, i.e. deep-set eyes, prominent low set ears, depressed nasal bridge with a broad nasal tip, small jaw, high arched palate and a pectus excavatum deformity of her chest. She had a history of poor growth (underweight for age and short stature). Her milestones were normal until 2.5 years of age. At the age of 5 years, she stopped walking and was assessed as having bilateral hip dysplasia. X-rays of her spine showed platyspondyly with anterior beaking of the vertebrae. X-rays of her hips showed small deformed capital femoral epiphyses and shallow dysplastic acetabular fossae. In the same year she became aphasic and developed a right hemiplegia. Her brain magnetic resonance imaging (MRI) showed a non-acute infarct in the left posterior parietal lobe and a more recent infarct in the right parieto-occipital lobe. A computerised tomography angiogram in the same year showed features of a vasculopathy involving the peripheral branches of the middle and posterior cerebral arteries.
At age 6 years she was diagnosed with nephrotic syndrome after she developed proteinuria, hypoalbuminaemia and hyper-cholesterolaemia. Her renal ultrasound demonstrated poor cortico-medullary differentiation. A renal biopsy showed focal segmental glomerulosclerosis. She had numerous admissions for recurrent relapses of the nephrotic syndrome. Her renal function remained normal during the course of her illness. She required recurrent Albumin infusions for the relapsing nephrotic syndrome. In addition, she was started on steroids and an angiotensin converting enzyme inhibitor. She developed osteoporosis and was commenced on bisphosphonates. She was not commenced on immunosuppressive therapy as her prognosis was deemed to be poor. She developed epilepsy that was treated with sodium valproate.
Later that year she had a recurrent stroke of the right cerebral hemisphere. A brain MRI in 2012 showed new infarcts in the right cerebral hemisphere (Fig. 1), cerebellum and left side of the pons and medulla. The magnetic resonance angiogram (MRA) showed a Moyamoya pattern (Fig. 2). The posterior cerebral arteries were now also occluded. She was assessed for a neurosurgical bypass procedure for the Moyamoya disease and was deemed to be a poor candidate owing to the extent of the brain lesions. She was investigated for other risk factors for recurrent strokes: protein C, protein S, antithrombin III and Factor V Leiden were normal. Her cardiac echogram was normal. Metabolic screens including urine for mucopolysacharidoses, as well as hexosaminidase A and B, and alpha-1-galactosidase were normal. Her serum lactate and ammonia levels were normal. Urine screen for aminoaciduria and organic acidaemias were also normal.
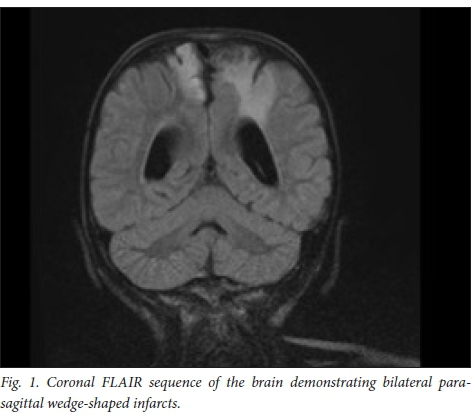
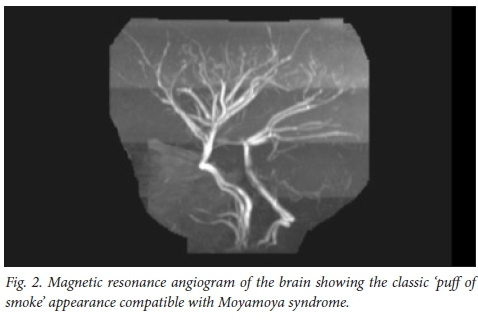
Genetic studies confirmed the presence of the c.1439 C>T mutation in the SMARCAL1 gene which has been described in patients with SIOD previously. She had no lymphopaenia or hypothyroidism. She died at the age of 7 years following an inter-current pneumonia.
Discussion
SIOD is caused by mutations in SMARCAL1 , which encodes a putative chromatin remodelling enzyme.[3] Mutations in SMARCAL1 gene results in loss of function of the SMARCAL1 protein and affects both cellular proliferation and differentiation and promotes vascular homoeostasis.[3] Genotype-phenotype correlation in SIOD is limited. The true prevalence is not known but is estimated at between 1:1 000 000 and 1:3 000 000 in North America. It is a pan-ethnic disorder.
Two forms of SIOD are described: a juvenile variant with adolescent onset of growth failure and renal dysfunction; and an infantile/ childhood-onset type that shows intrauterine growth restriction, followed by failure of linear growth in the first 2 - 3 years of life.[2] Our index patient showed features of the infantile/childhood-onset type.
SIOD should be suspected in a patient with the following clinical features:
• short stature secondary to a spondo-epiphyseal dysplasia
• steroid resistant nephrotic syndrome
• T Cell immunodeficiency resulting in recurrent infections
• characteristic facial features
• hyperpigmented macules on the skin.
Other less common clinical features include sparse hair, dental abnormalities, hypothyroidism and corneal opacities. Diagnosis is confirmed by identification of biallellic pathogenic variants in SMARCAL1 on molecular genetic testing. Descriptions of the neurological complications of SIOD are limited. One patient has been reported with white matter changes and four patients with cerebellar abnormalities.[4]
The association of vascular disease with SIOD was first noted by Spranger et al.,[5]who reported 4 patients with atherosclerotic cerebrovascular disease. In 1995, Ehrich et al.[6] described 3 further patients with recurrent strokes. Cerebral and cerebellar perfusion defects were demonstrated in these patients by positron emission tomography. Boerkoel[2] first described 2 patients with the classical Moyamoya pattern on MRI in 1998. Moyamoya disease is a chronic, occlusive cerebrovascular disease characterised by progressive stenosis at the terminal portion of the internal carotid artery and an abnormal vascular network at the base of the brain. It is postulated that a factor responsible for the production and maintenance of connective tissue is defective in SIOD. SMARCAL1 is expressed at low levels in vascular smooth muscle and endothelium. Mutations in SMARCAL1 would thus result in an intrinsic defect in the blood vessel wall that would predispose the vessel wall to fibroses.[7] Hypertension and hyperlipidemia caused by the nephrotic syndrome in SIOD may also accentuate an underlying defect in the arterial wall.[8] However, in the series of patients described by Erlich[5] the vascular disease progressed after successful renal transplantation suggesting that renal disease alone is not responsible for the cerebro-vascular disease.
The prognosis for those with childhood onset is poor. Most patients demise within the first decade of life.
Conclusion
Neurological complications in patients with SIOD are not uncommon.[9] Our patient presented with recurrent strokes and her neuroimaging confirmed Moyamoya syndrome. In our patient with SIOD and Moyamoya, disease the vasculopathy was progressive as described in previous case studies. She was not a candidate for a revascularisation procedure as she had extensive brain injury.
Acknowledgements. We acknowledge Dr Kuben Pillay, paediatric endocrinologist at Westville Hospital, who was the primary caregiver of the patient, made the diagnosis and reviewed the manuscript. We are grateful to Professor Neal Boerkoel and Marie Morimoto of the Department of Medical Genetics, University of British Columbia, Vancouver, British Columbia, Canada for performing the genetic studies as part of their research project.
Author contributions. Both authors were involved in the clinical care of the patient. RG was responsible for review of the literature and drafting of the manuscript. EN reviewed the manuscript.
Funding. None.
Conflicts of interest. None.
References
1. Schimke RN, Horton WA, King CR. Chondroitin-6-sulfaturia, defective cellular immunity, and nephrotic syndrome. Lancet 1971;298(7733):1088-1089. [ Links ]
2. Boerkoel CF, Nowaczyk MJM, Blaser SI, Meschino WS, Weksberg R. Schimke immune-osseous dysplasia complicated by Moyamoya phenomenon. Am J Med Genet 1998;78(2):118-122. [ Links ]
3. Boerkoel C F, Takashima H, John J, et al. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Gen 2002;30(2):215-220. https://doi.org/10.1038/ng821 [ Links ]
4. Lucke T, Clewing JM, Boerkoel CF, et al. Cerebellar atrophy in Schimke-immuno-osseous dysplasia. Am J Med Genet 2007;143A:2040-2045. https://doi.org/10.1002/ajmg.a.31878 [ Links ]
5. Spranger J, Hinkel GK, Stöss H, Thoenes W, Wargowski D, Zepp F. Schimke immuno-osseous dysplasia: A newly recognized multisystem disease. J Pediatr 1991;119:64-72. https://doi.org/10.1016/S0022-3476(05)81040-6. [ Links ]
6. Ehrich JHH, Burchert W, Schirg E, et al. Steroid resistant nephrotic syndrome associated with spondyloepiphyseal dysplasia, transient ischemic attacks and lymphopenia. Clin Nephrol 1995;43(2):89-95. [ Links ]
7. Clewing JM, Antalfy BC, Lücke T, et al. Schimke immuno-osseous dysplasia: A clinicopathological correlation. J Med Genet 2007;44(2):122-130. https://doi.org/10.1136/jmg.2006.04431310 [ Links ]
8. Lücke T, Marwedel KM, Kanzelmeyer NK, et al. Generalized atherosclerosis sparing the transplanted kidney in Schimke disease. Pediatr Nephrol 2004;19(6):672-675. https://doi.org/10.1007/s00467-004-1426-z. [ Links ]
9. Schmidt B, Christen HJ, Herkenrath P, Benz-Bohm G, Muller-Berghaus J, Querfeld U. Cerebral complications in Schimke immuno-osseous dysplasia. Eur J Pediatr 1997;156(10):789-791. [ Links ]
 Correspondence:
Correspondence:
Dr R Govender
govenderr2@ukzn.ac.za

