










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Child Health
On-line version ISSN 1999-7671
Print version ISSN 1994-3032
S. Afr. j. child health vol.12 n.2 Pretoria Apr./Jun. 2018
http://dx.doi.org/10.7196/sajch.2018.v12i2.1483
CASE REPORT
Neonatal haemolytic anaemia - a diagnostic approach to red cell membrane disorders
L SwartI; K NaidooII; E SchapkaitzIII; J PooleIV; T L CoetzerV
IMB ChB; Department of Molecular Medicine and Haematology, National Health Laboratory Service, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIMSc, PhD; Department of Molecular Medicine and Haematology, National Health Laboratory Service, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIIMB BCh, FCPath (SA), MMed (Haem); Department of Molecular Medicine and Haematology, National Health Laboratory Service, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IVBSc (Hons), PhD; Department of Paediatrics, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
VMB BCh, FCPaed (SA), DCH (SA); Department of Molecular Medicine and Haematology, National Health Laboratory Service, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
In neonates presenting with a non-immune haemolytic anaemia, a high index of suspicion is raised for hereditary red cell membrane disorders. The distinction between red cell membrane disorders, however, is often difficult in neonates in the absence of a complete family history. We describe a case of a 26-day-old female twin who presented with jaundice and severe haemolysis, which required multiple red cell transfusions. Laboratory investigations revealed a non-immune haemolysis. Red cell membrane extraction and sodium dodecyl sulphate-polyacrylamide gel electrophoresis analysis, including spectrin analysis, revealed the presence of mutant spectrin αI/74. A diagnosis of hereditary elliptocytosis with transient infantile poikilocytosis was favoured. On follow-up at 4 months, a decline in haemolysis was observed.
The timely diagnosis and management of neonatal haemolytic anaemia is of critical importance in neonatal units. In neonates presenting with hyperbilirubinaemia, non-immune haemolysis and a positive family history, a high index of suspicion is raised for hereditary red cell membrane disorders. These include hereditary spherocytosis (HS), hereditary elliptocytosis (HE) and hereditary stomatocytosis (HSt). These disorders can often be diagnosed by the characteristic red blood cell (RBC) shape on peripheral blood smear (PBS) (Fig. 1). However, the RBC shape is not always specific for the particular red cell membrane disorder. Appropriate specialised investigations of both the neonate and parents are indicated, with further management by a paediatric haematologist. We present a case of a 26-day-old female twin who presented with a persistent non-immune haemolytic anaemia, which required multiple red cell transfusions.
Case
A 26-day-old female neonate was referred to the Charlotte Maxeke Johannesburg Academic Hospital with a history of severe transfusion dependent haemolytic anaemia. Neonatal jaundice developed within 12 hours of delivery. At birth, the haemoglobin (Hb) was 10.9 g/dL and the unconjugated bilirubin was 240 μmol/L. This female dizygotic twin (birth weight 1 910 g) was delivered by caesarian section at 34 weeks' gestation for premature rupture of membranes. Both parents originated from Angola and an in vitro fertilisation pregnancy was performed with paternal sperm and a donor egg. As such, a complete family history could not be obtained.
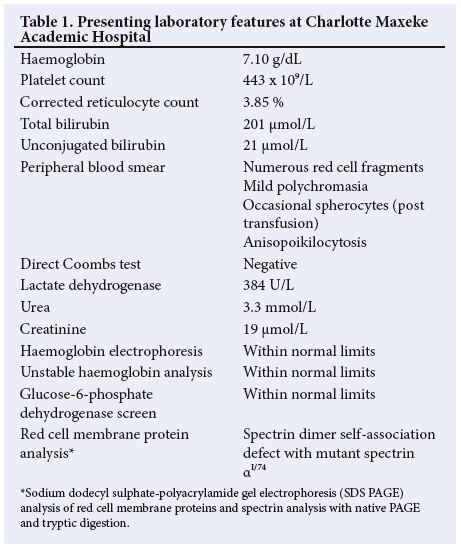
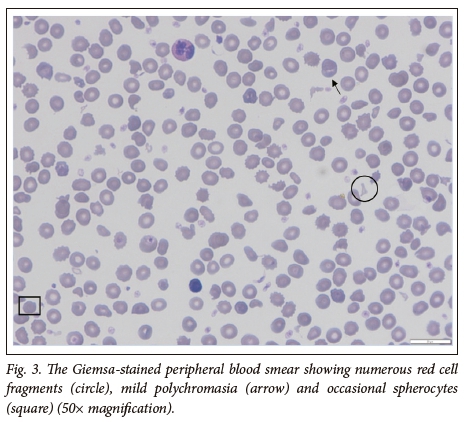
The female twin continued to haemolyse and was referred to our centre for specialist investigations and management (Fig. 2). The laboratory parameters are presented in Table 1. The Hb was 7.10g/dL on admission, with a corrected reticulocyte count of 3.85 %. The PBS showed moderate polychromasia and numerous red cell fragments, as well as occasional spherocytes (Fig. 3). The direct Coombs test was negative. Specialist investigations for non-immune haemolysis were performed, namely Hb electrophoresis, glucose-6-phosphate dehydrogenase screen and unstable Hb analysis, which were all within normal limits. Analysis of isolated red cell membrane proteins using sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS PAGE) analysis revealed no abnormalities. Native PAGE of isolated spectrin and structural analysis by limited digestion with trypsin indicated the presence of mutant spectrin αI. This favoured the diagnosis of HE with transient infantile poikilocytosis.
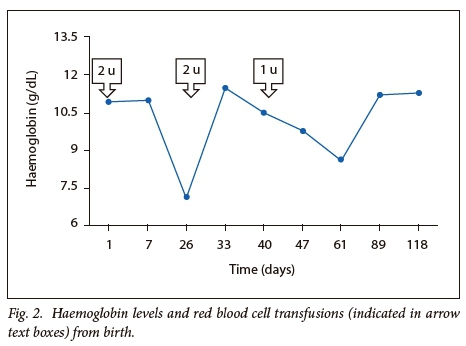
At four months of follow-up, a decline in haemolysis was observed with a Hb level of 11.3 g/dL in the female twin. The PBS showed mild polychromasia, occasional spherocytes and red cell fragments. The family returned to Angola and the patient was lost to follow-up. The male twin developed mild jaundice after birth which responded to phototherapy. He had no associated anaemia. Red cell membrane protein analyses, in the male twin, were not performed.
Discussion
HE is a hereditary red cell membrane disorder, which is characterised by mechanical instability of the red cell membrane skeleton.1The majority of the HE membrane defects occur in α and β spectrin,2 which result in disruption of the spectrin heterodimer self-association sites, impairing spectrin tetramer formation. This results in haemolysis.
HE is classified according to its clinical presentation into asymptomatic or non-haemolytic HE, HE with variable haemolysis, HE with transient infantile poikilocytosis and hereditary pyropoikiolcytosis (HPP).2 The clinical severity of HE described in family studies is markedly heterogenous.3 The most frequent presentation of HE is HE with variable haemolysis, which mostly follows a benign clinical course. This is typically inherited in an autosomal dominant fashion and the amount of mutant and normal spectrin are equivalent. In contrast, HE with transient infantile poikilocytosis and HPP present during the first year of life with severe transfusion dependent haemolytic anaemia. HPP is a severe autosomal recessive disorder. Cases of HPP show a marked spectrin dimer self-association defect and a decreased amount of spectrin. This severely weakens the membrane skeleton, resulting in red cell fragmentation and severe haemolysis. The distinction between HPP and HE with transient infantile poikilocytosis is often difficult in neonates presenting with severe haemolysis. Clinically, HPP cases typically remain transfusion dependent until a splenectomy is performed, in contrast to HE with transient infantile poikilocytosis which evolves into the mild HE phenotype.1-3 The diagnosis is confirmed by analysis of red cell membrane protein analysis, including spectrin analysis.
In this case, red cell membrane protein and spectrin analyses favoured the diagnosis of HE in association with transient infantile poikilocytosis. However, the FBC and PBS findings of a low mean cell volume (MCV), as a result of the significant red cell fragmentation and the absence of elliptocytes, suggested a diagnosis of HPP. Further, the clinical presentation was severe, with a requirement for multiple red cell transfusions. Since the proband had been transfused prior to performing specialist investigations, the presence of transfused red blood cells would have increased the relative amount of normal spectrin compared with the mutant spectrin. It is thus important to perform definitive diagnostic tests, such as red cell membrane protein studies prior to transfusion, in order to prevent diagnostic misinterpretations.
At ~4 months of age, there is a switch from fetal to adult Hb. This results in an increase in the stability of the red cell membrane skeleton. On follow-up, at 4 months of age in this case, there was a decline in haemolysis and the Hb was stable. The clinical and laboratory picture of HE usually emerges from ~18 months of age. A repeat spectrin analysis (performed at least 3 months post transfusion) is required to confirm the diagnosis. In addition, family studies are indicated. In members of the same family with this mutation, the clinical presentation may be heterogeneous with a phenotypically different disease. Unfortunately, the patient and her family have been lost to follow-up.
Conclusion
This case report emphasises the importance of reviewing the PBS routinely on all neonates presenting with anaemia and non-physiological jaundice. At a primary care level, initial laboratory investigations should include an FBC, PBS examination, reticulocyte count, direct Coombs test and a haemolytic workup. It should be noted that the classic morphologic abnormalities of HE (elliptocytes) are usually not present during the first few weeks of life. Specific investigations include SDS-PAGE analysis of red cell membrane proteins, native PAGE for spectrin analysis and molecular diagnostic testing if the clinical and laboratory findings suggest a hereditary red cell membrane disorder. It is important to perform the appropriate specialised laboratory tests prior to transfusion to avoid a delay in diagnosis.
Acknowledgements. None.
Author contributions. Writing: LS, KN, ES, TLC. Concept and design: LS, KN, ES, TLC. Analysis and processing: KN, TLC. Interpretation: KN, TLC, LS, ES. Treating paediatrician: JP.
Funding. None.
Conflicts of interest. None.
References
1. Coetzer TL. Erythrocyte membrane disorders. In: Williams Hematology. 9th ed. Kaushansky K, Lichtman MA, Prchal JT, et al. (eds). New York: McGraw Hill, 2016. [ Links ]
2. King MJ, Garcon L, Hoyer JD, et al. ICSH guidelines for the laboratory diagnosis of nonimmune hereditary red cell membrane disorders. Int J Lab Hematol 2015;37(3):304-325. https://doi.org/10.1111/ijlh.12335 [ Links ]
3. An X, Mohandas N. Disorders of red cell membrane. Br J Haematol 2008;141(3):367-375. https://doi.org/10.1111/j.1365-2141.2008.07091.x [ Links ]
 Correspondence:
Correspondence:
L Swart
leanne.swart@icloud.com
Accepted 31 January 2018


{kind=link}