










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Child Health
versão On-line ISSN 1999-7671
versão impressa ISSN 1994-3032
S. Afr. j. child health vol.11 no.3 Pretoria Out. 2017
http://dx.doi.org/10.7196/sajch.2017.v2i3.1300
RESEARCH
The clinical profile and outcome of children with West syndrome in KwaZulu-Natal Province, South Africa: A 10-year retrospective review
A KeshaveI; N Yende-ZumaII; L MubaiwaIII; M AdhikariIV
IMB ChB, MMed, FC Paed, Cert Paed Neuro; Department of Paediatric Neurology, Nelson R Mandela School of Medicine, University of KwaZulu-Natal, Durban, South Africa
IIBSc, BSc Hons, MSc; Centre for the AIDS Programme of Research in South Africa (CAPRISA), University of KwaZulu-Natal, Durban, South Africa
IIIMB ChB, FC Paed, MA Department of Paediatric Neurology, Nelson R Mandela School of Medicine, University of KwaZulu-Natal, Durban, South Africa
IVMB ChB, FC Paed, PhD;Department of Paediatric Neurology, Nelson R Mandela School of Medicine, University of KwaZulu-Natal, Durban, South Africa
ABSTRACT
BACKGROUND. West syndrome (WS) is a rare epileptic encephalopathy of infancy. There is currently no research on the incidence or prevalence of WS in Africa.
METHODS. We aimed to describe the outcome of children with WS at a quaternary-level hospital in KwaZulu-Natal, South Africa (SA). This was a retrospective chart review conducted on patients diagnosed with WS over a 10-year period. Eight children (males, n=7; African, n=6; Asian, n=2) identified with WS out of 2 206 admitted with epilepsy. The median age (range) at diagnosis was 7.5 (1 - 9) months. The average time between onset of epileptic spasms and diagnosis was 3.1 months.
RESULTS. Six patients had abnormal neuroimaging (atrophy (n=2); corpus callosum agenesis (n=2); tuberous sclerosis (n=1); focal dysplasia (n=1)). Drug management included sodium valproate (n=8), topiramate (n=7) and levetiracetam (n=3). Subsequent definitive treatment was intramuscular adrenocorticotrophic hormone (n=3), vigabatrin (n=2) and oral prednisone (n=4). Four (50%) patients had complete seizure remission (neuromigratory disorder (n=2); tuberous sclerosis (n=1); and idiopathic (n=1)) and 4 had partial remission (neonatal complications (n=3); idiopathic (n=1)).
DISCUSSION. Most of our patients had symptomatic WS, with 50% remission on treatment. Outcomes were poorer in our study when compared with those in published data.
CONCLUSION. Further collaborative studies are still needed to evaluate the true impact and prevalence of WS in SA.
West syndrome (WS) is a rare epileptic disorder of infancy or early childhood. Dr William James West first described the syndrome in 1841 when it affected his son.[1] It is an epileptic encephalopathy characterised by epileptic spasms, electroencephalographic (EEG) evidence of hypsarrhythmia and developmental delay or regression.[2] Infantile spasms involve the neck, trunk and extremities. Spasms are classified as either flexors (flexion of the arms, legs and neck with contraction of the abdominal muscles in a jack knife or salaam attack), extensors (extension of the neck and trunk with abduction or adduction of the arms and legs), or mixed flexor-extensor spasms (flexion of the neck, trunk and arms with extension of the legs).[3] Spasms usually appear in the first 2 years of life, with a peak incidence between 4 and 6 months, and sometimes continue until adolescence.[2] Hypsarrhythmia is described as highvoltage, random, slow and spike waves in the cortex that vary in duration and location, and occasionally may become generalised.[4]
The incidence of WS is well documented in developed countries, and a study in Finland reported an estimated incidence of 30.7 per 100 000 live births.[5] The incidence and prevalence of aetiological factors of WS in Africa are unknown.
Successful treatment of WS improves morbidity and mortality outcomes. In a long-term-outcome study of 214 Finnish children with WS over a 35-year period, a third of the patients died, of whom a third died before the age of 3 years.[6] Significant contributing factors to the poor prognosis are the lack of standardised treatment guidelines, and delays in commencing treatment. This was evident after the results of studies from the Pellock et al.[2]Lux et al,[1]Ito et al,[8]and Wilmshurst et al.[9] highlighted the treatment gap for WS. It was noted that further research was needed to formulate appropriate treatment protocols for WS with respect to pharmacological and non-pharmacological therapies, such as the ketogenic diet and neurosurgical interventions, such as vagal nerve stimulation, deep brain stimulation and epileptic surgery. There are numerous publications of studies regarding the classification, treatment, and prognosis of WS in developed countries.[10] Knowledge regarding the epidemiology and outcomes of children with WS in developing countries and in resource-constrained settings is limited.[11] There are no descriptive studies on WS in Africa. We hypothesised that the incidence would be higher and treatment outcomes poorer for WS in our setting than in developed countries. The aim of the study was to evaluate the clinical presentation and treatment outcomes of children with WS in KwaZulu-Natal Province, South Africa (SA), and to identify factors related to current practices in diagnosing and treating WS in the Paediatric Neurology Department at Inkosi Albert Luthuli Central Hospital.
Ethical approval was obtained from the Biomedical Research Ethics Committee of the University of KwaZulu-Natal (ref. no. BE419/15). Gatekeeper approval for use of the patients' records and data was obtained from the Department of Health and Inkosi Albert Luthuli Central Hospital.
Methods
The study was conducted at the Paediatric Neurology Department at Inkosi Albert Luthuli Central Hospital, a quaternary care centre in Durban, SA. The hospital serves the general population of KwaZulu-Natal. The patients are of mixed socioeconomic backgrounds.
Study design
Data were collected using a retrospective chart review of all patients diagnosed with WS over a 10-year period, from January 2005 to August 2015. Patients were identified from a data base of all patients <12 years of age admitted to the paediatric neurology ward with seizures or epilepsy. We identified 8 patients diagnosed with WS, from a total of 2 206 patients with seizures.
We extracted the following data from the patient files: sex, race, age of onset of infantile spasms, age at diagnosis, number of clusters per day, number of seizures per cluster, perinatal events, mode of delivery, retroviral status, aetiology, relation to tuberous sclerosis, anthropometry and developmental assessment, neuroimaging with magnetic resonance imaging (MRI) and electrophysiological studies with EEG.
Data on treatment were also captured, including the type of antiepileptic drug used, the maximum dose and side-effects, and the response to treatment. The outcome was determined by the degree of epileptic spasm reduction prior to discharge, and on follow-up. The response to treatment was categorised into three groups: complete spasm cessation, partial response (>50% reduction in spasms) and no response (<50% reduction in spasms). Information regarding seizure semiology, EEG findings, anthropometry and neurodevelopment that was recorded in patient files on clinic visits was also extracted.
Data analysis
Continuous data were summarised using means, and categorical data using proportions. Fisher's exact test was used to test for association between categorical variables. Statistical analyses were conducted using SAS version 9.4 (SAS Institute, USA).
Results
Demographics
The WS patients were predominantly male (87.5%, n=7). The majority (75%, n=6) were black African, and the others Indian (25%, n=2).
Perinatal history
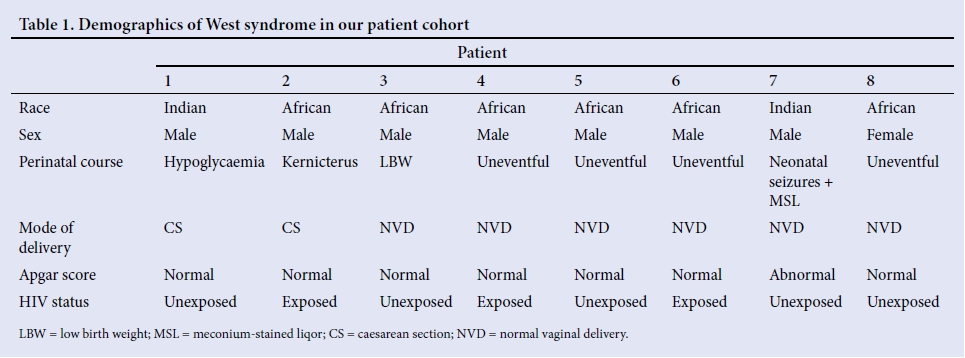
Table 1 shows the demographics of the perinatal history of the children in our patient cohort. Six patients (75%) were born by caesarean section. Four patients (50%) had abnormal perinatal courses. The remaining 4 patients had the following perinatal comorbidities: hypoglycaemia (n=1), kernicterus (n=1), neonatal seizures (n=1) and low birth weight (n=1). An abnormal Apgar score was defined as a score <6 at 5 minutes or 10 minutes, and this was noted in 1 patient (12.5%), who subsequently developed neonatal encephalopathy. Three patients (37.5%) were born to mothers infected with HIV, but were not infected themselves.
Presentation
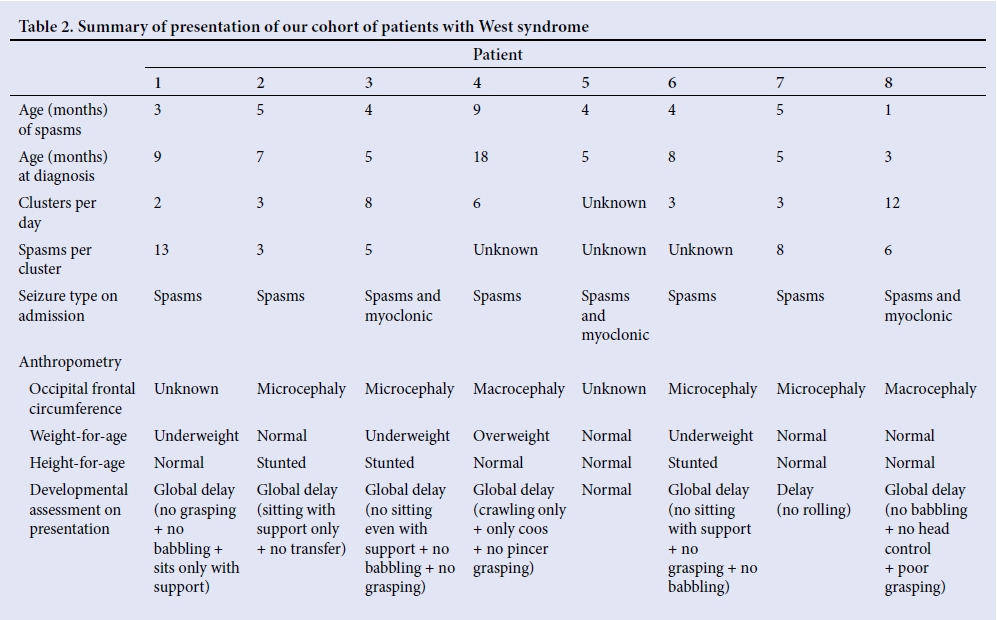
Table 2 shows a summary of the presentation of our patients with WS. The mean age of epileptic spasm onset was 4.4 (range 1 - 9) months, with the mean age at diagnosis being 7.5 (range 3 - 18) months. The average duration between onset of spasms and diagnosis was 3.1 months. Patients had on average 5 clusters per day, with 7 spasms per cluster. Three patients (37.5%) had mixed seizures, consisting of spasms and myoclonic seizures. A total of 66.7% (n=4) had microcephaly, with an occipital-frontal diameter <2 standard deviations (SD) for age, and 37.5% (n=3) were underweight for their age, and stunted, with length-for-age <2 SD. All patients presented with developmental delay, and the majority (n=6) had global developmental delay, affecting gross and fine motor skills (87.5% and 85.7%, respectively).
Investigations
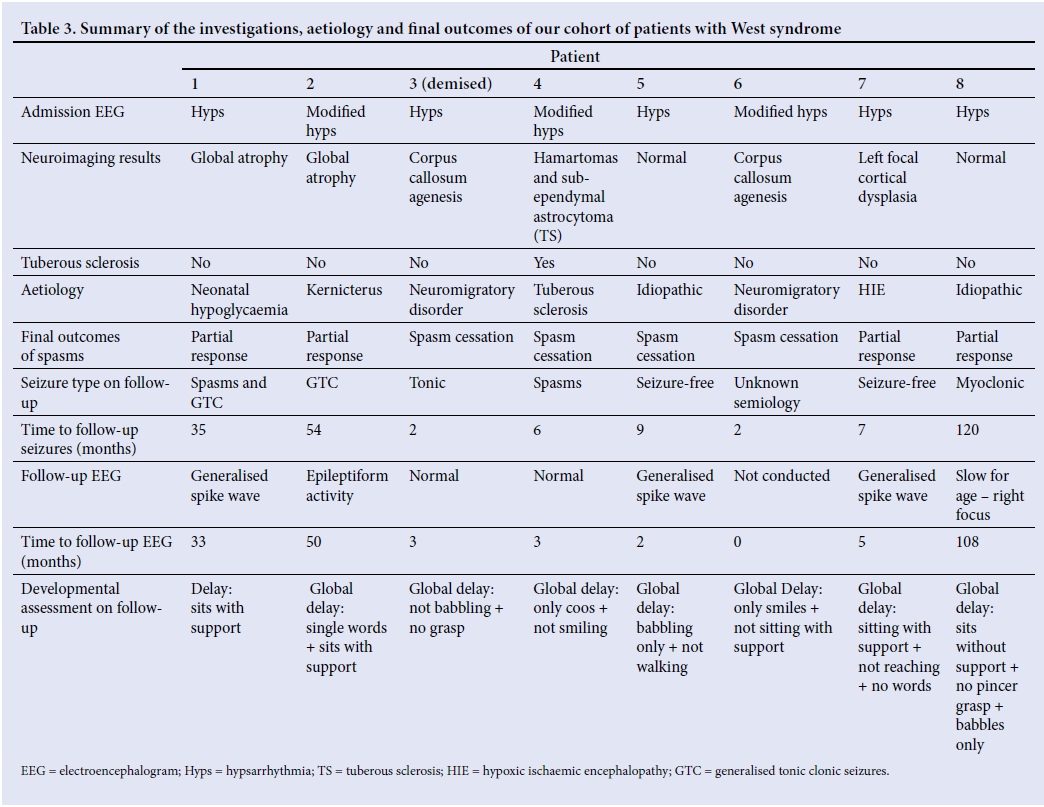
EEG findings on admission were modified hypsarrhythmia (37.5%, n=3) and classic hypsarrhythmia (62.5%, n=5). All patients had MRI of the brain on admission, and the findings were: normal (n= 2), global atrophy (n=2), corpus callosum agenesis (n=2), features of tuberous sclerosis (hamartoma and sub-ependymal giant cell astrocytoma) (n=1) and left focal cortical dysplasia (n=1). The causes associated with WS in our cohort of patients were cryptogenic (n=2), neuromigratory disorder (n=2), neonatal hypoglycaemia (n=1), kernicterus (n=1), tuberous sclerosis (n=1) and neonatal hypoxic ischaemic encephalopathy (n=1) (Table 3).
Treatment
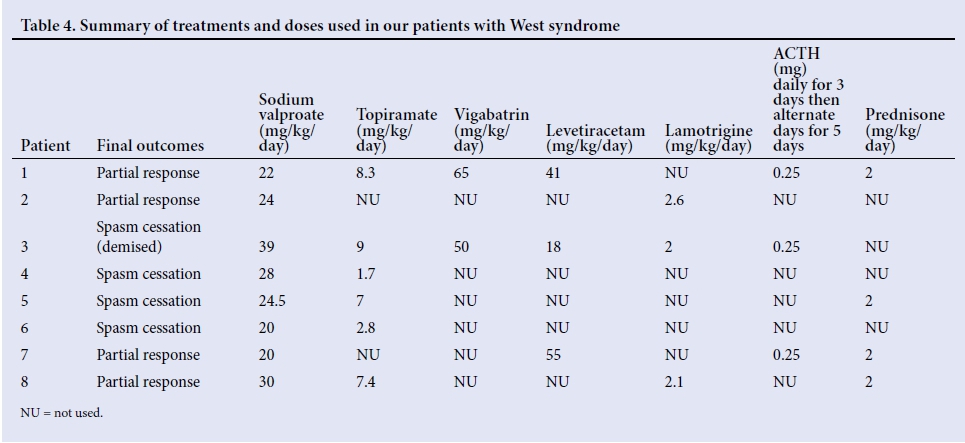
At our institution, the first-line therapy for patients presenting with suspected WS is sodium valproate. The second-line therapy is topiramate or lamotrigine, followed by escalation with sequential use of levetiracetam. Once a diagnosis of WS is confirmed, adrenocorticotropic hormone (ACTH) and vigabatrin are prescribed. Oral prednisone is used as an alternative if ACTH is not available. Patients are screened for tuberculosis, which is endemic in our population, prior to the use of ACTH and prednisone. None of our patients who received ACTH or prednisone had a positive screen for tuberculosis. A total of 11 drugs were used in different combinations for all 8 patients: sodium valproate (n=8), topiramate (n=7), clonazepam (n=5), prednisone (n=4), ACTH (n=3), levetiracetam (n=3), lamotrigine (n=3), vigabatrin (n=2), lorazepam (n=1), phenytoin (n=1) and phenobarbital (n=1) (Table 4).
Three patients had adverse reactions to sodium valproate and topiramate. The adverse effects of sodium valproate were hyperammonia (n=3) and thrombocytopenia (n=1). The adverse effects occurred with a prescribed average dose of sodium valproate of 44.7 mg/kg/day, and after a mean duration of treatment of 396 days. The main side-effect with topiramate was hypercarbia (n=1), with the average dose of topiramate being 9 mg/kg/day, and the average duration of treatment was 30 days. Only 1 patient in the study group died, as a result of comorbid sepsis and fulminant liver failure.
Final outcomes
An uneventful perinatal course was associated with a better outcome (75%), compared with the group that had an underlying perinatal event (25%). The mode of delivery and HIV status did not determine poor outcome. Outcome measures were: spasm cessation, cognitive development, epilepsy evolution, morbidity and mortality.
An earlier age of epileptic spasm presentation (<5 months) was associated with a better prognosis, with 60% of these children having spasm cessation, as compared with older children (>5 months), who had no spasm cessation (33.3%). There was no correlation between the age of diagnosis, head circumference, clusters per day and spasms per cluster with final outcomes. Only 1 patient had improved developmental milestones on follow-up. The patient, on presentation at 9 months old, was not able to grasp objects or babble, and sat only with support. At the follow-up visit at 35 months of age, the patient could speak full sentences, had a pen grip, could copy a circle and self-feed with a spoon. Patients who presented with modified hypsarrhythmia on EEG had slightly better outcomes with respect to seizure cessation than those with classic hypsarrhythmia (66.7% v. 40%). Neuroimaging findings did not correlate with prognosis.
A total of 37.5% (n=3) were prescribed ACTH, and 50% (n=4) had oral prednisone. The best treatment response, as indicated by spasm remission, was with vigabatrin (50%), followed by ACTH, lamotrigine and levetiracetam (33.3%). Our facility has access only to tetracosactide - a long-acting corticotrophin preparation administered intramuscularly.[12] Patients received 20 units (0.25 mg), which was appropriate for the age and weight of patients <1 year of age, daily for the first 3 days, and then on alternate days for 5 doses. A total of 14 days of treatment accounts for a single course. This low-dose regimen induced a good response to treatment, with spasm cessation in 2 out of 3 patients, and spasm cessation occurred within a week of commencing ACTH. One patient failed to respond owing to an incomplete course of ACTH, as the drug was not available. The oral dose for prednisone used was 2 mg/kg daily, tapered after 2 weeks. Four patients who received oral prednisone had the following outcomes: 2 were spasm-free, 1 had a partial response and 1 had no response. The favourable response was noted after a day of treatment.
Three patients who were not prescribed ACTH or prednisone were on dual therapy of sodium valproate and topiramate, and their outcomes were as follows: 2 had complete cessation of spasms, and 1 had a partial response. The patients with spasm cessation were both on sodium valproate and topiramate, while the 1 with partial response was on sodium valproate and lamotrigine.
Follow-up consultations
The patients with WS were regularly reviewed at the outpatients department after discharge, for an average of 29.4 (range 2 - 120) months. Patients presented with the following seizures: spasms (n=1), generalised tonic clonic (n=2), myoclonic (n=1), mixed seizures (n=1), i.e. both spasms and myoclonic seizures, and seizure-free (n=2). The patients with WS showed improvement in gross and fine motor skills on clinical assessment, but a worsening of language and social skills on follow-up. The EEG findings on follow-up, after a median (range) of 25.5 (2 - 108) months were: normal (n=2), general spike wave (n=3), generalised epileptiform activity (n=1), slow for age and right focus (n=1).
Discussion
This retrospective study describes the profile and outcomes of patients with WS, who accounted for 0.4% of all patients admitted with seizures at our institution. There was a male predominance, in keeping with studies in Sweden and Iceland that showed a 1:3 female to male ratio.'13,141 Patients who had a normal perinatal course had a better outcome, with 75% having spasm remission, as compared with 25% with an abnormal perinatal course. The cause of WS related to a perinatal insult was 50% in our study population. This is higher than that documented in studies from developed countries, but is similar to that found in other developing countries.[7,11] We also noted that there was no correlation between HIV exposure and outcome.
The mean age of onset and diagnosis of WS was similar to that in both developed and developing countries, where a peak incidence of epileptic spasms occurs between 3 and 7 months.[2, 11] The average length of time between spasm onset and diagnosis was 3.1 months. This was greater than that in developed countries' which showed a lag time of 25 - 45 days, but less than in other developing countries, at 7.9 months.[11, 15] There was no correlation between the number of spasms per cluster or clusters per day and final neurological outcome in our study. The number of spasms at presentation was similar to that recorded in other studies.[4]
All our patients had resolution of hypsarrhythmia when assessed on follow-up visits. Two patients had normal EEG pattern, and 6 patients had evolution to other epileptiform activities. The natural progression of hypsarrhythmia is to resolve with brain maturation.[16] Evolution of hypsarrhythmia to other interictal patterns has been reported, and was noted in our study.[4]
In our study, 2 patients had seizure remission on follow-up consultation, and 2 had persistent infantile spasms. The remainder had evolved to other seizure types. Spasms in WS rarely persist after 5 years of age. Fifty percent of epileptic spasms cease at ~3 years of age. In 60% of patients with WS, the spasms may evolve into other seizure types.[17] Studies show that 27% of patients with WS evolve to Lennox-Gastaut syndrome (LGS), and that 40% of patients with LGS had WS.[18] This evolution of WS to LGS was not evident in our study, since the follow-up period of patients was rather short, and we had few patients.
Results of published studies have revealed that 70% of patients with WS have symptomatic WS and an identifiable aetiology on neuroimaging.'191 This was similar in our study, with 6/8 patients having an underlying cause. Previous studies have shown that patients with identified normal neuroimaging have a better clinical outcome.[20] However, this was not evident in our study, which showed a 50% partial response to spasms in patients with a normal MRI.
The 2010 US Consensus Report on infantile spams concluded that treatment with steroids was beneficial.[2] The overall response rate in our study was in keeping with other studies[2,9] that recommended the use of ACTH and prednisone for treatment of spasms in WS. The patient who did not respond to prednisone had focal cortical dysplasia on MRI. None of our patients who received either ACTH or prednisone developed side-effects to the drugs. This was not in keeping with studies that showed an 85% incidence of adverse effects in WS patients treated with steroids, with an increased risk at higher doses (maximum doses of prednisone 60 mg/day and ACTH 60 IU on alternate days). The adverse effects reported included increased appetite, irritability, hypertension and glycosuria.[7,21] The low dose used in our patients possibly minimised the adverse effects.
Vigabatrin was prescribed for 2 patients, and there was a 50% cessation of spasms. This was in keeping with a placebo-controlled trial that showed a 68% spasm cessation rate.[22] Vigabatrin has been shown to be the treatment of choice in patients with tuberous sclerosis as a cause for WS. Several studies have demonstrated that patients with tuberous sclerosis treated with vigabatrin had complete remission of spasms.[2,9,23] Vigabatrin has not been found to be superior to ACTH or prednisone. Vigabatrin is not the first-line therapy in WS patients without tuberous sclerosis, because of the associated adverse effects of irreversible retinal dysfunction and concentric visual-field constriction.[9,24]
The treatment-algorithm strategy in our institution for WS was initiation with sodium valproate, followed by adjunctive therapy of topiramate or lamotrigine, and escalation to levetiracetam until a definitive diagnosis of WS is made. Studies on the use of sodium valproate for WS show inconsistent efficacious outcomes. The current literature shows a positive response rate of 73% to sodium valproate therapy within 2 weeks.[25] Topiramate as add-on therapy was used in an open-label study in 11 patients with refractory WS.'261 The results of the study showed that 50% had spasm cessation, and an additional 4 patients showed a 50% reduction in spasm frequency.
Research evidence on the use of levetiracetam is limited, and inconclusive on its efficacy as monotherapy in WS.[27,28] In our study, we found that all 3 patients treated with levetiracetam continued to have seizures, and were subsequently treated with ACTH.
Study limitations
The limitations of the study are that this was a retrospective study with a limited number of patients from a single centre. We found a male predominance, with a higher incidence of perinatal comorbidity, in our cohort of patients. Epileptic spasms had an earlier age of onset, with a longer duration from onset of spasms to time of diagnosis, as compared with developed countries. This can be attributed to the poor clinical identification of WS, and hence the delay in referral to our centre for further investigations and treatment. Most of our patients had an underlying diagnosis and abnormal neuroimaging, with neuromigratory disorders being the most common finding. Half of our patients responded to our treatment algorithm.
Lack of access to ACTH for treatment limited the chances of a favourable outcome in our patients. Only 2 patients (25%) remained seizure-free on follow-up, compared with studies from the USA (46%)'291 and the UK (75%).'71 The mortality rate was 12.5% in this study.
Conclusion
WS is a rare epileptiform encephalopathy, and a high index of suspicion is needed for identification. This is the first published study to describe the clinical profile and outcome of children with WS in Africa. The diagnosis of WS is usually delayed, owing to late referral to specialist care and lack of resources, and treatment options are limited by the availability of medication. Prospective studies with larger patient numbers are required to evaluate better treatment practices and outcome measures in Africa. This will allow for the formulation of standardised treatment guidelines for resource-poor settings, and improve the outcomes of children with WS.
Acknowledgments. The Department of Paediatric Neurology medical staff at Inkosi Albert Luthuli Central Hospital for the management of the patients.
Author contributions. Data collection, extraction and drafting of manuscript was done by AK. LM and MA supervised the project, and read and corrected the manuscript. Data analysis was performed by NYZ, a biostatistician. Drafts were then read and approved by all authors.
Funding. None.
Conflict of interest. None.
References
1. West WJ. On a peculiar form of infantile convulsions. Letter to the editor. Lancet 1841;1:724-725. [ Links ]
2. Pellock JM, Hrachovy R, Shinnar S, et al. Infantile spasms: A US consensus report. Epilepsia 2010;51(10):2175-2189. https://doi.org/10.1111/j.1528-1167.2010.02738.x [ Links ]
3. Kellaway P, Hrachovy RA, Frost JD Jr, Zion T. Precise characterization and quantification of infantile spasms. Ann Neurol 1979;6(3):214-218. https://doi.org/10.1002/ana.410060306 [ Links ]
4. Hrachovy RA, Frost JD Jr. Infantile epileptic encephalopathy with hypsarrhythmia (infantile spasms/West syndrome). J Clin Neurophysiol 2003;20(6):408-425. https://doi.org/10.1097/00004691-200311000-00004 [ Links ]
5. Riikonen R. Epidemiological data of West syndrome in Finland. Brain Dev 2001;23(7):539-541. https://doi.org/10.1016/s0387-7604(01)00263-7 [ Links ]
6. Riikonen R. Long-term outcome of patients with West syndrome. Brain Dev 2001;23(7):683-687. https://doi.org/10.1016/s0387-7604(01)00307-2 [ Links ]
7. Lux AL, Edwards SW, Hancock E, et al. The United Kingdom Infantile Spasms Study (UKISS) comparing hormone treatment with vigabatrin on developmental and epilepsy outcomes to age 14 months: A multicentre randomised trial. Lancet Neurol 2005;4(11):712-717. https://doi.org/10.1016/s1474-4422(05)70199-x [ Links ]
8. Ito M, Seki T, Takuma Y. Current therapy for West syndrome in Japan. J Child Neurol 2000;15(6):424-428. https://doi.org/10.1177/088307380001500615 [ Links ]
9. Wilmshurst JM, Gaillard WD, Vinayan KP, et al. Summary of recommendations for the management of infantile seizures: Task Force Report for the ILAE Commission of Pediatrics. Epilepsia 2015;56(8):1185-1197. https://doi.org/10.1111/epi.13057 [ Links ]
10. Young C. National survey of West syndrome in Taiwan. Brain Dev 2001;23(7):570-574. https://doi.org/10.1016/s0387-7604(01)00271-6 [ Links ]
11. Kaushik JS, Patra B, Sharma S, Yadav D, Aneja S. Clinical spectrum and treatment outcome of West Syndrome in children from Northern India. Seizure 2013;22(8):617-621. https://doi.org/10.1016/j.seizure.2013.04.014 [ Links ]
12. Hamano S, Yamashita S, Tanaka M, Yoshinari S, Minamitani M, Eto Y. Therapeutic efficacy and adverse effects of adrenocorticotropic hormone therapy in West Syndrome: Differences in dosage of adrenocorticotropic hormone, onset of age, and cause. J Pediatr 2006;148(4):485-488. https://doi.org/10.1016/j.jpeds.2005.11.041 [ Links ]
13. Sidenvall R, Eeg-Olofsson O. Epidemiology of infantile spasms in Sweden. Epilepsia 1995;36(6):572-574. https://doi.org/10.1111/j.1528-1157.1995.tb02569.x [ Links ]
14. LuoAvigsson P, Olafsson E, Sigurthardottir S, Hauser WA. Epidemiologic features of infantile spasms in Iceland. Epilepsia 1994;35(4):802-805. https://doi.org/10.1111/j.1528-1157.1994.tb02514.x [ Links ]
15. Lagae L, Verhelst H, Ceulemans B, et al. Treatment and long term outcome in West syndrome: The clinical reality. A multicentre follow up study. Seizure 2010;19(3):159-164. https://doi.org/10.1016/j.seizure.2010.01.008 [ Links ]
16. Wong M, Trevathan E. Infantile spasms. Pediatric Neurol 2001;24(2):89-98. https://doi.org/10.1016/s0887-8994(00)00238-1 [ Links ]
17. Riikonen R. A long-term follow-up study of 214 children with the syndrome of infantile spasms. Neuropediatrics 1982;13(1):14-23. https://doi.org/10.1055/s-2008-1059590 [ Links ]
18. Rantala H, Putkonen T. Occurrence, outcome, and prognostic factors of infantile spasms and Lennox-Gastaut syndrome. Epilepsia 1999;40(3):286-289.https://doi.org/10.1111/j.1528-1157.1999.tb00705.x [ Links ]
19. Wirrell EC, Shellhaas RA, Joshi C, et al. How should children with West syndrome be efficiently and accurately investigated? Results from the National Infantile Spasms Consortium. Epilepsia 2015;56(4):617-625. https://doi.org/10.1111/epi.12951 [ Links ]
20. Saltik S, Kocer N, Dervent A. Informative value of magnetic resonance imaging and EEG in the prognosis of infantile spasms. Epilepsia 2002;43(3):246-252.https://doi.org/10.1046/j.1528-1157.2002.14001.x [ Links ]
21. Riikonen R, Donner M. ACTH therapy in infantile spasms: Side effects. Arch Dis Child 1980;55(9):664-672. https://doi.org/10.1136/adc.55.9.664 [ Links ]
22. Appleton RE, Peters AC, Mumford JP, Shaw DE. Randomised, placebo- controlled study of vigabatrin as first-line treatment of infantile spasms. Epilepsia 1999;40(11):1627-1633. https://doi.org/10.1111/j.1528-1157.1999.tb02049.x [ Links ]
23. Granstrom ML, Gaily E, Liukkonen E. Treatment of infantile spasms: Results of a population-based study with vigabatrin as the first drug for spasms. Epilepsia 1999;40(4):950-957. https://doi.org/10.1111/j.1528-1157.1999.tb00802.x [ Links ]
24. Koul R, Chacko A, Ganesh A, Bulusu S, Al Riyami K. Vigabatrin associated retinal dysfunction in children with epilepsy. Arch Dis Child 2001;85(6):469-473. https://doi.org/10.1136/adc.85.6.469 [ Links ]
25. Siemes H, Spohr HL, Michael T, Nau H. Therapy of infantile spasms with valproate: Results of a prospective study. Epilepsia 1988;29(5):553-560. https://doi.org/10.1111/j.1528-1157.1988.tb03760.x [ Links ]
26. Glauser TA, Clark PO, Strawsburg R. A pilot study of topiramate in the treatment of infantile spasms. Epilepsia 1998;39(12):1324-1328. https://doi.org/10.1111/j.1528-1157.1998.tb01331.x [ Links ]
27. Gümü§ H, Kumanda§ S, Per H. Levetiracetam monotherapy in newly diagnosed cryptogenic West syndrome. Pediatr Neurol 2007;37(5):350-353. https://doi.org/10.1016/j.pediatrneurol.2007.06.019 [ Links ]
28. Mahmoud AA, Rizk TM, Mansy AA, Ali JA, Al-Tannir MA. Ineffectiveness of topiramate and levetiracetam in infantile spasms non-responsive to steroids. Open labeled randomised prospective study. Neuroscience 2013;18(2):143-146. https://doi.org/10.1016/j.jns.2013.07.2038 [ Links ]
29. Knupp KG, Coryell J, Nickels KC, et al. Response to treatment in a prospective national infantile spasms cohort. Ann Neurol 2016;79(3):475-484. https://doi.org/10.1002/ana.24594 [ Links ]
 Correspondence:
Correspondence:
A Keshave
amith.keshave@gmail.com


{kind=link}
{kind=link}
{kind=link}
{kind=link}