










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Child Health
versão On-line ISSN 1999-7671
versão impressa ISSN 1994-3032
S. Afr. j. child health vol.11 no.2 Pretoria Jun. 2017
http://dx.doi.org/10.7196/SAJCH.2017.v11i2.1277
CASE REPORT
Chediak-Higashi syndrome presenting in the accelerated phase
S PalaniyandiI; E SivaprakasamII; U PasupathyIII; L RavichandranIV; A RajendranV; F R SumanVI; S Rajendra PrasadVII
IMD; Department of Paediatrics, Sri Ramachandra University, Chennai, India
IIDCH, DNBDepartment of Paediatrics, Sri Ramachandra University, Chennai, India
IIIMD Department of Paediatrics, Sri Ramachandra University, Chennai, India
IVDCH, DNB Department of Paediatrics, Sri Ramachandra University, Chennai, India
VMD, DM Department of Paediatrics, Sri Ramachandra University, Chennai, India
VIMD; Department of Pathology, Sri Ramachandra University, Chennai, India
VIIDCH Department of Paediatrics, Sri Ramachandra University, Chennai, India
ABSTRACT
Chediak-Higashi syndrome (CHS) is an extremely rare autosomal recessive disorder characterised by recurrent pyogenic infections, partial oculocutaneous albinism, and mild bleeding. The most reliable finding that helps in diagnosis is abnormally large granules in leukocytes and other granule-containing cells. Herein we report a case of CHS in a 3-month-old girl who presented to us in the accelerated phase of the disease. The case is reported because of the extreme rarity of CHS presenting in the accelerated phase at diagnosis.
A 3-month-old female child, the third child born of a second-degree consanguineous marriage and developmentally normal with a normal birth history, presented to us with complaints of fever for 3 days. There was a history of hypopigmentation over the skin for the past month, with a history of recurrent respiratory tract infections. There was no family history of similar complaints.
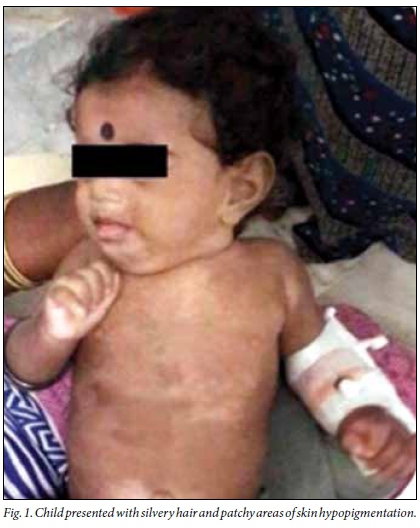
Examination revealed an active, anthropometrically normal, febrile child with axillary lymphadenopathy and hepatosplenomegaly. The child had silvery hair with patchy areas of skin hypopigmentation over the face, trunk and limbs (Fig. 1). Other systems were normal.
Evaluation showed bicytopenia (low haemoglobin of 6.6 g/dL and platelets of 60 000/μL) with elevated serum triglyceride (323 mg/dL) and ferritin (2 734 ng/mL) - features suggestive of haemophagocytic lymphohistiocytosis (HLH). A familial or infectious cause was suspected. Blood and urine cultures were sterile. Toxoplasmosis, rubella, cytomegalovirus, herpes simplex and HIV (TORCH) screening was also negative. Ultrasonography of the abdomen revealed mild ascites. Radiographs of the chest and skull were normal.
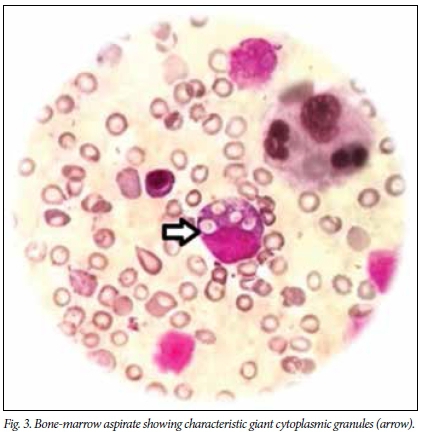
Peripheral smear (Fig. 2) and bone-marrow aspirate (Fig. 3) showed characteristic giant cytoplasmic granules suggestive of Chediak-Higashi syndrome (CHS). Skin biopsy (Fig. 4) revealed irregularly placed giant melanosomes, consistent with CHS. Ophthalmic evaluation was normal. Ebstein-Barr virus work-up (IgG and IgM) was negative. CHS presenting with features of HLH was taken as the accelerated phase. Genetic testing for CHS1/LYST gene mutations is not available in India, and therefore this test could not be performed. T-cell subset levels were not done because of financial constraints.


The child was treated with intravenous piperacillin, tazobactam, and amikacin, which were later changed to meropenem and fluconazole owing to persistent high-grade fever spikes. Initiation of chemotherapy was planned as per the HLH 2004 protocol,[1] and a bone-marrow transplant was to be considered; however, the parents did not opt for any escalation of treatment and wanted to continue with the conservative management. The child was discharged at their request and succumbed to her illness 2 days after discharge.
Discussion
CHS is a rare disease that follows an autosomal recessive pattern of inheritance. Less than 500 cases have been reported worldwide. The first case in India was diagnosed in 1982.[2]
Genetic studies suggest a mutation in the lysosomal trafficking regulator (CHS1/LYST) gene located at 1q42, resulting in abnormal organellar protein trafficking and aberrant fusion of vesicles, further resulting in a failure to transport lysosomes to the appropriate site of action.[3] Altered lysosomes/granules are found in all cell types in CHS, and are the hallmark of the disease.
CHS can be suspected on presentation of partial oculocutaneous albinism with a history of recurrent infections.[3]Staphylococcus aureus is the predominant cause while Streptococcus pyogenes and Pneumococcus spp. are the other common infectious organisms. Most of the cases also present with leukopenia, thrombocytopenia and coagulopathy. Photosensitivity has been reported in many cases. Differential diagnosis includes Hermansky-Pudlak syndrome and Griscelli syndrome.[4,5] Hermansky-Pudlak syndrome, an autosomal recessive disorder, is characterised by partial oculocutaneous albinism and a platelet storage pool deficiency. Griscelli syndrome is another autosomal recessive disorder that is characterised by similar partial oculocutaneous albinism with immunodeficiency, which is attributed to a mutation in one of three intracellular trafficking genes.
Defective neutrophil chemotaxis, degranulation and bactericidal activity could be the reason for recurrent infections. Pathological aggregation and uneven distribution of melanosomes play a role in hypopigmentation. The degree of hypopigmentation varies. The hair can be light blonde, grey or white with a metallic sheen. In darkly pigmented races, hypopigmentation is appreciated more in sun-exposed areas. Iris and retinal pigmentation is also reduced; light-coloured eyes are seen. Impaired platelet aggregation may contribute to the mild bleeding diathesis found in some cases. The mild coagulation defect in such cases can result in easy bruising and abnormal bleeding, especially noted in mucosal tissue.
Diagnosis can be made with a simple peripheral smear for the classic giant azurophilic granules, which are peroxidase-positive, in all granule-containing cells including the peripheral blood and bone marrow.[6] Giant melanosomes can be seen on skin melanocytes. Genetic testing for CHS1/LYST gene mutations can confirm the diagnosis. This gene is large with most mutations being unique, and therefore identifying the exact mutation is a challenge. Prenatal diagnosis is made by amniocentesis or chorionic villus sampling for enlarged lysosomes - this helps in early diagnosis and treatment before the accelerated phase.[7]
There are two phases in the progress of the disease: a stable or chronic phase; and a progressive or accelerated phase. The stable phase is characterised by recurrent infections. This phase can be managed with appropriate use of antibiotics or antifungal agents with adequate hygiene. About 10% of patients survive early childhood despite serious infections, but develop severe, debilitating neurological manifestations such as mental retardation, peripheral neuropathy and seizures in adolescence and early adulthood.
The accelerated phase may occur soon after birth, as in our case, or years later, and is usually fatal unless intervention occurs rapidly. It mimics a lymphoma-like scenario and is considered a form of familial HLH.[8] Epstein-Barr virus and a lack of natural killer cell function has been implicated in the accelerated phase.[9] There is lymphohistiocytic infiltration of virtually all organs with more profound immune deficiency. Affected individuals present with fever, increased hepatosplenomegaly and lymphadenopathy, with worsening pancytopenia and bleeding.[10] Other presentations can include unexplained hepatosplenomegaly and unexplained neurological abnormalities, especially in an older child, in the form of ataxia, tremors, muscle weakness, sensory loss, cranial nerve palsies, progressive intellectual decline and seizures. Movement disorders, such as Parkinson's disease and dementia can also occur.
The accelerated phase is treated with chemotherapy as per the HLH 2004 protocol. Haematopoietic stem cell transplant is the ultimate treatment for the immunological and haematological manifestations of CHS.[11,12] However, it has no effect on the neurological symptoms and oculocutaneous albinism.
Successful transplantation depends on having an HLA-identical donor. HLA-non-identical transplant remains an experimental approach. There has been a report of successful bone-marrow transplantation in a 2-year-old male with CHS in the accelerated phase with hereditary elliptocytosis, the boy being clinically well post transplant.[13]
Most patients with CHS die in their first decade if a stem-cell transplant is not done, although patients have been reported as old as 27 years.[14] In a study of 35 children with CHS, the 5-year prognosis post transplantation was 62%. Prognosis is better if the transplant is done before the onset of the accelerated phase. Atypical presentation of CHS can include subtle or absent oculocutaneous albinism, insignificant or less frequent infections, subtle bleeding manifestations and progressive neurological findings that are highly variable and nonspecific.[15]
A carefully examined peripheral smear can clinch the diagnosis. Early detection facilitates early bone-marrow transplant, which is the only curative approach for CHS.
Acknowledgements. None.
Author contributions. All authors were involved in the management of the patient. SP drafted the manuscript. UP, LR, and SRP reviewed the manuscript. AR and FRS provided haematolgy and pathology expertise, respectively. ES critically reviewed the manuscript.
Funding. None.
Conflict of interest. None.
References
1. Henter J-I, Horne AC, Arico M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007,48(2):124-131. https://doi.org/10.1002/pbc.21039 [ Links ]
2. Seth P, Bhargava M, Kalra V. Chediak-Higashi syndrome. Indian Paediatr 1982;19(11):950-952. [ Links ]
3. Kaplan J, De Demenico I, Ward DM. Chediak-Higashi syndrome. Curr Opin Hematol 2008;15(1):22-29. https://doi.org/10.1097/moh.0b013e3282f2bcce [ Links ]
4. Diukman R, Tangiawara S, Cowan MJ, Golbus MS. Prenatal diagnosis of Chediak-Higashi syndrome. Prenat Diagnosis 1992;12(11):877-885. https://doi.org/10.1002/pd.1970121105 [ Links ]
5. De Saint Basile G. Chediak-Higashi and Griscelli syndromes. Immunol Allergy Clin North Am 2002;22(2):301-317. [ Links ]
6. Burkhardt JK, Wiebel FA, Hester S, Argon Y. The giant organelles in beige and Chediak-Higashi fibroblasts are derived from late endosomes and mature lysosomes. J Exp Med 1993;178(6):1845-1856. https://doi.org/10.1084/jem.178.6.1845 [ Links ]
7. Valente NY, Machado MC, Boggio P, et al. Polarised light microscopy of hair shafts aids in the differential diagnosis of Chediak-Higashi and Griscelli- Prunieras syndrome. Clinics 2006;61(4):327-332. https://doi.org/10.1590/s1807-59322006000400009 [ Links ]
8. Nargund AR, Madhumathi DS, Premalatha CS, Rao CR, Appaji L, Lakshmidevi V. Accelerated phase of Chediak-Higashi syndrome mimicking lymphoma: A case report. J Pediat Hematol Oncol 2010;32(6):e223-e226. https://doi.org/10.1097/mph.0b013e3181e62663 [ Links ]
9. Merino F, Henle W, Ramirez-Duque P. Chronic active Epstein Barr virus infection in patients with Chediak-Higashi Syndrome. J Clin Immunol 1986;6(4):299-305. https://doi.org/10.1007/bf00917330 [ Links ]
10. Scherber E, Beutel K, Ganschow R, Schulz A, Janka G, Stadt U. Molecular analysis and clinical aspects of four patients with Chédiak-Higashi syndrome (CHS). Clin Genet 2009;76(4):409-412. https://doi.org/10.1111/j.1399-0004.2009.01205.x [ Links ]
11. Ayas M, Al-Ghonaium A. In patients with Chediak-Higashi syndrome undergoing allogenic SCT, does adding etoposide to the conditioning regimen improve the outcome of bone marrow transplant? Bone Marrow Transplant 2007;40(6):603. http://dx.doi.org/10.1038/sj.bmt.1705774. [ Links ]
12. Eapen M, DeLaat CA, Baker KS, et al. Hematopoietic cell transplantation for Chediak Higashi syndrome. Bone Marrow Transpl 2007;39(7):411-415. https://doi.org/10.1038/sj.bmt.1705600 [ Links ]
13. Islam AS, Hawsawi ZM, Islam MS, Ibrahim OA. Chédiak-Higashi syndrome: An accelerated phase with hereditary elliptocytosis. Ann Saudi Med 2001; 21(3-4):221-224. https://doi.org/10.5144/0256-4947.2001.221 [ Links ]
14. Gallin JI, Elin RJ, Hubert RT, et al. Efficacy of ascorbic acid in Chediak-Higashi syndrome: Studies in humans and mice. Blood 1979;53:226-234. [ Links ]
15. Karim MA, Suzuki K, Fukai K, et.al. Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak-Higashi syndrome. Am J Med Genet 2002;108(1):16-22. https://doi.org/10.1002/ajmg.10184 [ Links ]
 Correspondence:
Correspondence:
E Sivaprakasam
layaped@gmail.com

