










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Child Health
On-line version ISSN 1999-7671
Print version ISSN 1994-3032
S. Afr. j. child health vol.9 n.1 Pretoria Jan. 2015
CASE REPORT
Biochemical and genetic diagnosis of Smith-Lemli-Opitz syndrome in South Africa
G A E SolomonI; G JonesII; G de JongIII; A D MaraisIV
IMSc; Chemical Pathology, Clinical Laboratory Sciences, University of Cape Town, South Africa; Cape Heart Group, Medical Research Council of South Africa, Cape Town, South Africa
IIMB ChB, FCP (SA) (Paed); Sandton Mediclinic, Johannesburg, South Africa
IIIMB ChB, BSc Hons (Genetics), MMed (Paed), MD; Department of Molecular Biology and Human Genetics, Faculty of Medicine and Health Sciences, Tygerberg Hospital, Stellenbosch University, Cape Town, South Africa
IVMB ChB, FCP (SA); Chemical Pathology, Clinical Laboratory Sciences, University of Cape Town, South Africa; Cape Heart Group, Medical Research Council of South Africa, Cape Town, South Africa; National Health Laboratory Service, Groote Schuur Hospital, Cape Town, South Africa
ABSTRACT
BACKGROUND: The Smith-Lemli-Opitz syndrome (SLOS), due to defective function of 7-dehydrocholesterol reductase, is an autosomal recessive disorder that is more common than other defects in cholesterol biosynthesis. The dysmorphology can be suggestive, but biochemical and genetic investigations are required for confirmation of this diagnosis to assist with the management of the patient and planning for future children in affected families. Objective. To perform biochemical and genetic work-ups in four South African families of European ancestry with suspected SLOS in a range of presentations, from early fatality, congenital malformations, feeding problems and developmental delay
METHODS: Plasma was analysed by ultraviolet spectrophotometry. The genetic cause was investigated by polymerase chain reaction, followed by high-resolution melting and sequencing of amplicons displaying abnormal patterns
RESULTS: Spectrophotometry confirmed the diagnosis in three families. Genetic confirmation was made in these patients, and carrier status confirmed in the parents of the fatal case. All the patients were of European ancestry, and the mutations reflected those in European studies
CONCLUSION: This rare disorder should be considered in antenatal assessment when increased nuchal lucency is detected on sonography, or in newborns with syndactyly, hypotonia and feeding problems. Less severe forms could present with developmental delay and behavioural problems. Confirmation of the diagnosis may assist in decisions about nutritional management as well as future pregnancies in the affected family and primary relatives
The clinical description of a dysmorphic syndrome by Smith, Lemli and Opitz in 1964[1] was linked with abnormal sterol biosynthesis 30 years later.[2] A systematic review[3] of the effects of abnormal cholesterol biosynthesis indicated that the plasma accumulates 7-dehydrocholesterol and its precursor, and is often associated with hypocholesterolaemia. Nevertheless, the cholesterol concentration may be in the normal range and should not be a deterrent for further investigation when the clinical features are suggestive of this syndrome. Faecal and biliary sterols are abnormal, and bile acid metabolism is also affected. Porter[4,5] reviewed cholesterol biosynthetic errors. Cholesterol precursors that accumulate in Smith-Lemli-Opitz syndrome (SLOS), lathosterolosis and desmosterolosis disrupt intrauterine development at a critical period of facial and brain development, when cholesterol binds covalently to sonic hedgehog protein involved in signalling to responding cells.[6] Engel-king[7] investigated the pathogenesis in a mouse model, and demonstrated that the precursors were operative in bringing about the abnormalities rather than the lack of cholesterol, as suppression of the production of precursors by lovastatin ameliorated the condition.
A clinical review[8] indicated the prevalence of SLOS to be between 1 in 20 000 - 70 000 births in individuals of European descent, with a gene frequency of ~1% for a mutation in 7-dehydrocholesterol reductase (7DHCR). The clinical spectrum of manifestations is broad, but can be summarised as craniofacial abnormalities including cleft palate and microcephaly, postaxial polydactyly and syndactyly of the toes, cardiac defects, pyloric stenosis, aganglionosis of the colon and ambiguous genitalia. Failure to thrive, hypotonia and developmental delay as well as self-injurious behaviour and autism are reported. Photosensitivity may be marked. The manifestations depend on the severity of the disruption of enzyme activity, and compensatory mechanisms may vary as well. The spectrum has been subdivided into the severe extreme (type 2) and the less severe (type 1); the latter may not be clinically recognised until developmental delay or behavioural problems are noticed. The fetal manifestations have been documented.[9] The diagnosis is best made by gas chromatography and mass spectrometry, but ultraviolet spectrophotometry can detect 7-dehydrocholesterol in extracts from plasma;[10] however, other abnormal precursor sterols could be overlooked by this method. Dried blood spot analysis may not be ideal, as 7-dehydrocholesterol is prone to oxidation.[11] Amniotic fluid can be used to confirm the diagnosis antenatally.[12]
The genetic causes of SLOS in Europe have been summarised[13] and compared with those in the USA. There were some differences in distribution in the European countries, but by far the most common mutation (35%) was at the intron-exon junction of exon 8, with p.trp151X (10%), p.thr95met (8%) and p.val326leu (7%). The first two mutations may have appeared ~3 000 years ago in north-west and north-east Europe. It is not clear whether the heterozygous state confers an advantage, but 7-dehydrocholesterol is the precursor to forming vitamin D in the skin.
Information about SLOS is limited in South Africa (SA). A diagnosis on clinical grounds has been reported.[14,15] In this article, we report experience with referred cases and wish to make medical practitioners aware of this condition in order to improve diagnosis, antenatal diagnosis and genetic counselling, as well as to ensure optimal management.
Methods
Blood samples (anticoagulated with ethylene diamine tetra-acetate) were accepted from referring medical practitioners who suspected SLOS on clinical grounds, including samples from the parents, with fasting lipid profiles when possible. Consent for genetic diagnosis was obtained by the medical practitioners. A brief description is given for each family in order of referral. The information is summarised in Table 1 along with the results of genetic investigation.
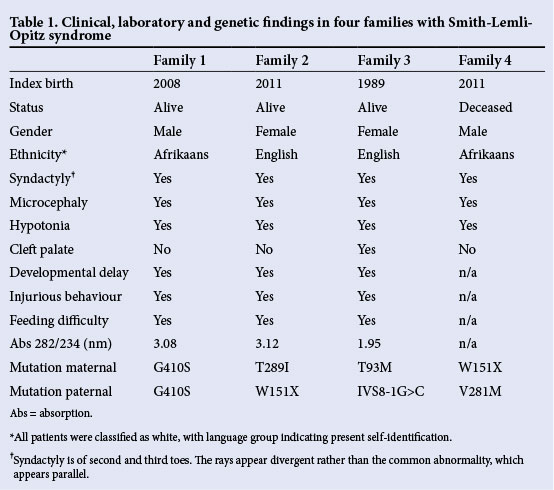
Family 1
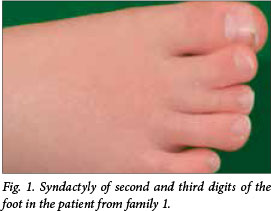
The first-born boy to unrelated parents had abnormal nuchal translucency by sonographic examination at 12 weeks of gestation. Later, short femora and a ventricular septal defect were identified. At birth by caesarean section in 2008, microcephaly, hypospadias and syndactyly (Fig. 1) were noted along with hypotonia and poor sucking reflexes. Neurological development was impaired and feeding difficulties were encountered. Nasogastric feeding was undertaken and later a gastrostomy was performed for feeding. The plasma cholesterol concentration was 1.1 - 1.5 mmol/L. Cholesterol intake was increased, and plasma cholesterol rose to 3.5 mmol/L. The impression was that such feeding improved wellbeing and behaviour. The raised plasma 7-dehydrocholesterol (750 μmol/L) was confirmed by ultraviolet spectrophotometry. Transaminase elevation was noted and considered to be part of the inherited disorder. At the age of 4 years, his mass is 22 kg and his height is 0.98 m. He can indicate his wishes but cannot speak. He lives at home and attends a schooling facility during the day.
Family 2
The first-born girl of unrelated healthy parents had an unremarkable antenatal course, but at caesarean section in 2011 was noted to be hypotonic, microcephalic, had bifrontal narrowing, small palpebral fissures, anteverted nares, syndactyly on the feet and mild deformities of the hand. Feeding difficulties included poor swallowing, reflux and vomiting. A gastrostomy was performed to assist with feeding at 4 months and a Nissen fundoplication was performed to deal with reflux. The biochemical diagnosis of SLOS was made at the age of
6 months, with a 7-dehydrocholesterol concentration of 836 μmol/L. With dietetic help, cholesterol supplementation was undertaken with a synthetic nutritional formula; staff involved in her treatment were of the opinion that she improved. The plasma cholesterol concentrations ranged from 3.3 to 4.1 mmol/L on cholesterol supplementation. On combination treatment with simvastatin (0.3 mg/kg/day), plasma 7-dehydrocholesterol was 0.125 μmol/L. Head banging, finger biting and pulling of hair started at 14 months. Investigations for ataxia revealed abnormalities of the cerebellar vermis on nuclear magnetic resonance imaging. At the age of 2 years, her developmental assessment placed her at 1 year and she had begun developing autistic behaviour.
Family 3
The first daughter was born with defects and died without a diagnosis within hours, in 1987. The second daughter, born in 1989, had multiple abnormalities including microcephaly, syndactyly and no nasal bridge. She also had a cleft palate. Tube feeding was instituted and projectile vomiting was noted. The cleft palate was repaired after a year, but feeding remained problematic. She required positioning so that liquid diets could drain into the stomach. Repeated ear infections resulted in admissions to hospital. Self-injurious behaviour was noted early on, necessitating the use of a protective helmet. She did eventually lose sight in both eyes owing to injury. At the age of 24 years, she has a height of 1.13 m and a mass of 22 kg. She cannot speak, is unable to sit on her own, sleeps little and is incontinent. When sedation was required, diazepam and most other commonly used agents failed, but hydroxyzine was suitable. The third child does not have SLOS.
Family 4
The first boy of unrelated healthy parents was born with a mass of 2.38 kg and low Apgar scores. There were multiple defects and he died on the second day. The antenatal course, including sonography, was unremarkable. At birth, the following features were recorded: webbed neck, rockerbottom feet, polydactyly of the hands and feet, and cardiac murmur. The clinical diagnosis of SLOS was suggested. The next pregnancy terminated spontaneously, but thereafter a normal female child was born. The diagnosis was pursued in order to counsel the primary relatives, as two brothers had married two sisters.
Biochemical diagnosis
Plasma was separated and sterols were extracted into hexane.[10] Absorbances were recorded at 234 nm and 282 nm, and the ratio was calculated. The molar extinction coefficient was determined with commercially available 7-dehydrocholesterol. The presence of 7-dehydrocholesterol was confirmed by argentation thin-layer chromatography.
Genetic diagnosis
Genomic DNA was extracted using the QIAamp DNA Mini Kit from Qiagen (USA) and screened for mutations in the DHCR7 gene using quantitative real-time polymerase chain reaction (PCR) high-resolution melting (HRM) (RT-qPCR-HRM).
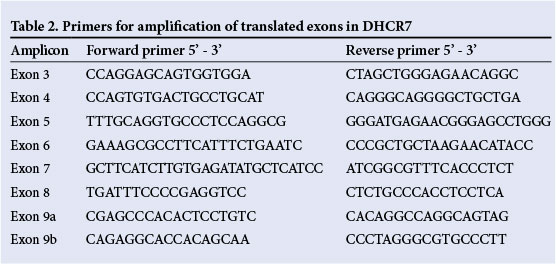
Primer pairs were designed to amplify the coding exons 3 - 9 of the DHCR7 gene, with amplicon lengths ranging from 160 to 370 base pairs (Table 2). Primer melting temperatures were adjusted for the LCGreen family of double-stranded DNA binding dyes. Each exon was analysed in a single PCR product, except exon 9, which was split into two amplicons. Samples were amplified on the LightScanner32 (Idaho Technology Inc, USA), an HRM-enabled, real-time PCR machine, and screened for sequence variations by curve analysis in a single run.
PCR was performed in a 10 μL reaction volume containing 50 ng of genomic DNA, 0.5 μΜ of each primer, 1x (undiluted) LightScanner Master Mix and PCR-grade water to adjust to the total volume of 10 μL. If necessary, 0.5% dimethyl sulphoxide (DMSO) was added to the reaction. The reaction conditions included an activation step at 95oC for 2 minutes, followed by 40 cycles of 95oC for 10 seconds, variable melting temperatures for 20 seconds, and 72oC for 20 seconds. Before the HRM step, products were denatured at 95oC for 5 seconds and renatured at 40oC for 30 seconds. HRM was carried out over the range of 60oC to 95oC, with a ramp rate of 0.3oC/s.
Results
The spectrophotometric analysis proved effective for the diagnosis of SLOS, as the ratio of absorbances at 234 nm and 282 nm was unequivocally raised in the patients when compared with a large number of control plasma samples. The patients from families 1, 2 and 3 had ratios of 3.08, 3.12 and 1.95, respectively (normal ratio <0.30).
The mutations identified were all previously reported to be pathogenic, and only one patient was a true homozygote for a pathogenic mutation. The mutations and their association with the parents are indicated in Table 1. The mutations were: exon 4 (threonine substituted by methionine at amino acid 93, T93M), exon 6 (tryptophan codon results in a stop of translation, W151X), exon 8 (threonine substituted by isoleucine at amino acid 289, T289I) and at the junction of the 8th intron and 9th exon, IVS8-1).
Discussion
Healthcare in SA is constrained by limited resources, and the focus is on primary healthcare, including antenatal care, childbirth and health of the infant. The emphasis on the most common disorders is appropriate, but patients with uncommon or severe disorders, especially those that develop complications competing for facilities and resources, should at least have a diagnosis for making decisions about management. Not only is there a need for assessment of these patients by gynaecologists, obstetricians and paediatricians, but also by clinical geneticists. Special biochemical and genetic investigations that are essential for a diagnosis are limited in SA. We describe four cases in whom the most common severe cholesterol biosynthetic error was fully characterised in families of European ancestry. Not unexpectedly, the mutations correspond with the most frequently reported mutations in Europe, and the clinical manifestations are similar.
The prevalence of SLOS as well as the carrier frequency of this recessive disorder is not well studied in populations other than those of European descent. The clinical features of SLOS are somewhat nonspecific and can vary. The dysmorphology may be less striking with age. The features that could indicate SLOS during pregnancy are increased nuchal lucency and the presence of various malformations. Diagnosis can be made on the amniotic fluid, which has been studied for the presence of 7-and/or 8-dehydrocholesterol and its ratio to total cholesterol.[12,16] The expression of the disorder is not only related to the residual enzyme activity but also to certain modifying factors. The clinical appearance at birth is suggestive but not specific, as other disorders need to be considered because several clinical features are shared, especially two other cholesterol biosynthetic errors (desmosterolosis, lathosterolosis). A careful evaluation should differentiate SLOS from Williams syndrome, WolfHirschhorn syndrome, Smith-Magenis syndrome, Malpuech syndrome, Rett syndrome, craniofacial ciliopathies as well as disorders that can render male genitalia ambiguous. Feeding difficulties and failure to thrive may arise in the neonatal and infantile period. Nutritional support in this period may require special care for adequate energy and nutrient intake, and mode of delivery: nasogastric tube feeding may suffice but percutaneous gastric feeding may be required. Confirmation of the diagnosis indicates that the child will continue failing to thrive, and will need more aggressive intervention for feeding. It is uncertain whether a cholesterol-enriched diet improves development, as the brain relies on de novo synthesis of cholesterol. Simvastatin, as an inhibitor of hydroxy-methylglutaryl co-enzyme A reductase, will limit the production of both 7-dehydrocholesterol and cholesterol, with the former building up as a result of the defect in this disorder. If there is residual activity to synthesise cholesterol but a reduction of its precursor that may adversely affect its function(s), then there may be improvement. There has, however, been a cautionary note about this therapy.[18] Developmental delay, behavioural problems and autism may later lead to the identification of SLOS. Moreover, termination of pregnancy may be elected when there is an antenatal diagnosis of the disorder, especially in those with a severe phenotype. Proving the diagnosis may thus enhance clinical decisions on management, minimise hospitalisation and reduce expenses for unnecessary investigations, of which some may be very expensive.
In Europe, the most common four mutations overall account for 60% of SLOS. These four mutations were identified in the first four families of European ancestry in SA in whom the diagnosis of SLOS was made. This rare disorder appears not to have a prominent founder effect in SA, as there is a range of mutations. Furthermore, there are three mutations in the two families of Afrikaner ancestry. The parents of the child in family 1 traced back their family without convergence until at least 1740. Systematic genetic detection of mutations in 7DHCR in the population at large is not warranted owing to the rarity of the condition, but consideration should be given to this disorder if antenatal development is abnormal. Biochemical confirmation of the diagnosis of SLOS without genetic substantiation will allow correct counselling about the risk of the condition being present in 25% of future pregnancies. Genetic identification will allow detection of carriers in the parents' siblings and other relatives, whose offspring should be monitored more closely, as the frequency for mutations in 7DHCR is suspected in about 1% of European populations.
The formal diagnosis of SLOS by gas chromatography and mass spectrometry is not available in SA. Such diagnostic investigation proved unaffordable in overseas laboratories, as these and other costs in management are supported to only a limited extent by medical schemes, and state hospitals and clinics and their laboratory services. However, the spectrophotometric test is reliable and affordable, and has been shown to be suitable for amniotic fluid extracts with confirmation on thin-layer chromatography. In the case of SLOS, the concurrence of syndactyly, hypotonia, feeding difficulties and other malformations is highly suggestive of the diagnosis. Ideally, the index of suspicion for severe inherited metabolic disorders should be raised in order that referral and investigation will lead to diagnosis and optimal management amid limited resources. Specialised centres in which clinics work in conjunction with specialised laboratories should be supported to provide for the increasing need to diagnose and manage metabolic disorders that are amenable to biochemical and/or genetic diagnosis. Once the mutations have been identified, chorionic villus biopsy can be undertaken to identify the disorder at the 11th week of pregnancy. Not only would such centres assist in reducing the burden of SLOS within the affected families, but they would also provide important information for teaching and planning of healthcare in SA.
Acknowledgments. We would like to thank Mr AR Mohamed for performing screening tests, Ms B Ratanjee for assistance with sample preparation, Dr M Witsch-Baumgartner and Prof. G Utermann for confirmation of the findings of the first patient, and the parents for assistance in the work-up of families.
References
1. Smith DW, Lemli L, Opitz JM. A newly organized syndrome of multiple congenital abnormalities. J Pediatr 1964;64(2):210-217. [http://dx.doi.org/10.1016/S0022-3476(64)80264-X] [ Links ]
2. Opitz JM, de la Cruz F. Cholesterol metabolism in the RSH/Smith-Lemli-Opitz syndrome: Summary of an NICHD conference. Am J Med Genet 1994;50(4):326-338. [ Links ]
3. Salen G, Shefer S, Batta AK, et al. Abnormal cholesterol biosynthesis in the Smith-Lemli-Opitz syndrome. J Lipid Res 1996;37:1169-1180. [ Links ]
4. Porter FD. Malformation syndromes due to inborn errors of cholesterol synthesis. J Clin Invest 2002;110(6):715-724. [http://dx.doi.org/10.1172/JCI16386] [ Links ]
5. Porter FD, Herman GE. Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res 2011;52(1):6-34. [http://dx.doi.org/10.1194/jlr.R009548] [ Links ]
6. Porter FD. Cholesterol precursors and facial clefting. J Clin Invest 2006;116(9):2322-2325. [http://dx.doi.org/10.1172/JCI29872] [ Links ]
7. Engelking LJ. Severe facial clefting in Insig-deficient mouse embryos caused by serol accumulation and reversed by lovastatin. J Clin Invest 2006;116(9):2356-2365. [http://dx.doi.org/10.1172/JCI28988] [ Links ]
8. Porter FD. Smith-Lemli-Opitz syndrome: Pathogenesis, diagnosis and management. Eur J Hum Genet 2008;16(5):535-541. [http://dx.doi.org/10.1038/ejhg.2008.10] [ Links ]
9. Quilin C, Loget P, Verloes A, et al. Phenotypic spectrum of fetal Smith-Lemli-Opitz syndrome. Eur J Med Genet 2012;55(2) :81 -90. [http://dx.doi.org/10.1016/j.ejmg.2011.12.002] [ Links ]
10. Honda A, Batta AK, Salen G, Tint GS, Chen TS, Shefer S. Screening for abnormal cholesterol biosynthesis in the Smith-Lemli-Opitz syndrome: Rapid determination of plasma 7-dehydrocholesterol by ultraviolet spectroscopy. Am J Med Genet 1997;68(3):288-293. [ Links ]
11. Gelzo M, Dello Russo A, Corso G. Stability study of dehydrocholesterols in dried spot of blood from patients with Smith-Lemli-Opitz syndrome, using filter-paper treated with butylated hydroxytoluene. Clin Chim Acta 2012;413(3-4):525-526. [http://dx.doi.org/10.1016/j.cca.2011.11.008] [ Links ]
12. Griffiths WJ, Wang Y, Karu K, et al. Potential of sterol analysis by liquid chromatography-tandem mass spectrometry for the prenatal diagnosis of Smith-Lemli-Opitz syndrome. Clin Chem 2008;54(8):1317-1324.[http://dx.doi.org/10.1373/clinchem.2007.100644] [ Links ]
13. Witsch-Baumgartner M, Schwentner I, Gruber M, et al. Age and origin of major Smith-Lemli-Opitz syndrome (SLOS) mutations in European populations. J Med Genet 2008;45(4):200-209. [http://dx.doi.org/10.1136/jmg.2007.053520] [ Links ]
14. De Jong G, Kirby PA, Muller LM. RSH Smith Lemli Opitz Syndrome: Severe phenotype with ectrodactyly. Am J Med Genet 1998;75(3):283-287. [ Links ]
15. Moodliar M, Singh R. Smith-Lemli-Opitz syndrome. Paediatric Quarterly 2010;2:17-20. [ Links ]
16. Kelley RI. Diagnosis of Smith-Lemli-Opitz syndrome by gas chromatography/ mass spectrometry of 7-dehydrocholesterol in plasma, amniotic fluid and cultured fibroblasts. Clin Chim Acta 1995;236(1):45-48. [ Links ]
17. Starck L, Lvvgren-Sandblom A, Bjvrkhem I. Simvastatin in Smith Lemli Opitz Syndrome: A safe approach? Am J Med Genet 2002;113(2):183-189. [http://dx.doi.org/10.1002/ajmg.10722] [ Links ]
 Correspondence:
Correspondence:
A D Marais
david.marais@uct.ac.za

