










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Child Health
versión On-line ISSN 1999-7671
versión impresa ISSN 1994-3032
S. Afr. j. child health vol.8 no.4 Pretoria nov. 2014
http://dx.doi.org/10.7196/SAJCH.655
SHORT REPORTS
Atypical teratoid/rhabdoid tumour in a supratentorial location: A report of two cases
N MahomedI; J NaidooII; S DlangamandlaV; S AndronikouVI; S PatherIII; K PillayIV
IMB BCh, FCRad (Diag) (SA), MMed, DHN Fellow (Boston); Department of Diagnostic Radiology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIMB BCh, FCRad (Diag) (SA), Dip Paed Rad; Department of Diagnostic Radiology, Faculty of Health Sciences, University of Cape Town, South Africa
IIIMB BCh, FCPath (SA), MMed Anat Path; Department of Anatomical Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IVMB ChB, FCPath (SA), FRCPath (UK), MMed Anat Path; Division of Anatomical Pathology, Faculty of Health Sciences, University of Cape Town, South Africa
VMB ChB, FCRad (Diag) (SA); Department of Diagnostic Radiology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
VIMB BCh, FCRad (Diag) (SA), FRCR, PhD; Department of Diagnostic Radiology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
Atypical teratoid/rhabdoid tumour of the central nervous system is a rare, highly aggressive childhood malignancy. The age of presentation is usually <2 years, but this tumour may occur in other age groups. The typical location is the posterior fossa, with supratentorial origin less common. We present two cases of atypical teratoid/rhabdoid tumours, with the suprasellar location of one case proving to be a diagnostic radiological challenge.
Atypical teratoid/rhabdoid tumour of the central nervous system (CNS) is a rare, highly aggressive childhood malignancy.[1, 2] The prognosis is generally poor.[3, 4] The typical location is the posterior fossa; less than a third occur supratentorially,[5] and most are off the midline.[1] We present two patients with supratentorial location (one suprasellar, which is very unusual), both demonstrating a peripheral rim of restricted diffusion on magnetic resonance imaging (MRI), an important diagnostic feature of atypical teratoid/rhabdoid tumours.[1, 2, 4]
Case 1
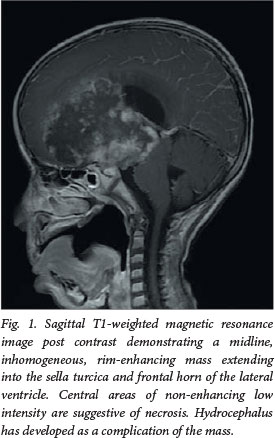
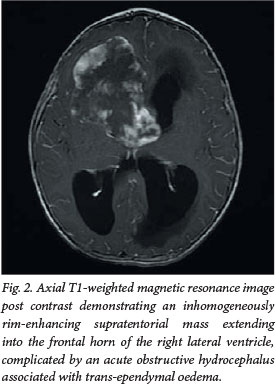
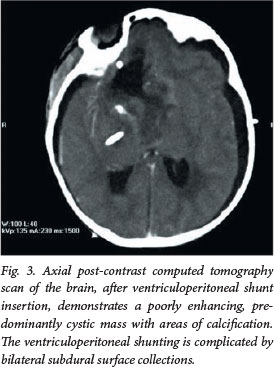
A 3-month-old girl presented to our institution with a history of convulsions. Postcontrast MRI demonstrated an inhomogeneously enhancing, centrally necrotic suprasellar mass, which extended into the frontal horn of the lateral ventricle, complicated by obstructive hydrocephalus (Figs 1 and 2). Diffusion-weighted imaging (DWI)/apparent diffusion coefficient (ADC) mapping demonstrated a peripheral ring of restricted diffusion within the tumour. 'Blooming' of areas on gradient echo was confirmed as calcification on a computed tomography (CT) scan performed after ventriculoperitoneal shunt insertion (Fig. 3). No spinal metastases were demonstrated on MRI.
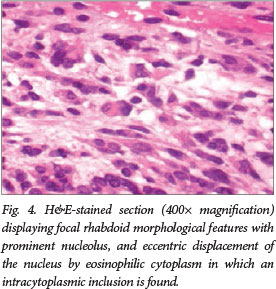
Pathological assessment confirmed the presence of a high-grade malignant neoplasm with foci of coagulative tumour necrosis and frequent mitotic figures. The architecture of the tumour varied from well-defined tumour cell nests to haphazardly distributed spindle cells. Some tumour cells displayed distinct rhabdoid morphology (Fig. 4) characterised by abundant eosinophilic cytoplasm with intracytoplasmic inclusions, eccentrically displaced vesicular nuclei and prominent nucleoli. Histopathological diagnosis of an atypical teratoid/rhabdoid tumour (World Health Organization (WHO) grade IV) was confirmed by diffuse immunoreactivity for vimentin (an intermediate filament), focal cytoplasmic and membrane staining with epithelial membrane antigen (EMA), and focal staining with smooth-muscle actin (SMA). Ki-67 immunohistochemistry revealed a high proliferation index of ~ 50%.
Glial fibrillary acidic protein (GFAP), neurofilament, cytokeratin and desmin immunohistochemistry were negative. Immunohistochemistry for germ cell markers was also negative.
The patient became comatose and died shortly after ventriculoperitoneal shunting, prior to any definitive treatment.
Case 2
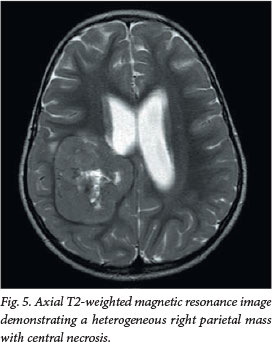
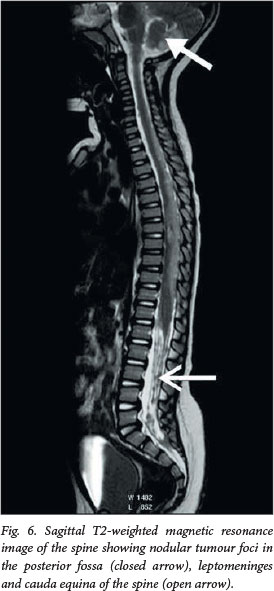
A 2-year-old girl presented with convulsions and new-onset left-sided weakness. MRI revealed a heterogeneous mass with central necrosis in the right parietal lobe, causing a mass effect on the right lateral ventricle and complicated by obstructive hydrocephalus (Fig. 5). The mass enhanced heterogeneously post contrast, with a central necrotic component. A peripheral rim of restricted diffusion was demonstrated on DWI/ADC mapping. CSF seeding of the tumour was detected along the ventral surface of the brainstem and on the cerebellopontine angle cisterns, the anterolateral temporal lobes and posterior fossa, the leptomeninges and the cauda equina of the spine (Fig. 6).
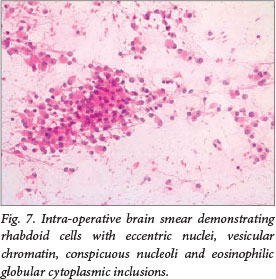
An intra-operative diagnosis of atypical teratoid/rhabdoid tumour was made based on the morphology of the brain smears. The cells had round eccentric nuclei, conspicuous nucleoli, abundant eosinophilic cytoplasm and paranuclear inclusions (Fig. 7). Paraffin sections confirmed the diagnosis and showed prominent vessels, perivascular pseudorosettes and areas of dystrophic calcification. On immunohistochemical staining, the tumour was strongly positive for vimentin, GFAP and EMA. The final pathological diagnosis was an atypical teratoid/rhabdoid tumour (WHO grade IV).
The patient died 2 days after biopsy and insertion of a ventriculoperitoneal shunt.
Discussion
Atypical teratoid/rhabdoid tumour of the CNS is a rare, highly aggressive childhood malignancy.[1, 2] The age of presentation is usually <2 years, but the tumour may occur in other age groups.[1-3]Prognosis is generally poor, with a 5-year survival rate of 28%.[3, 4] The typical location is the posterior fossa (62%), while only 27% of these tumours occur supratentorially.[5] Atypical teratoid/rhabdoid tumours usually occur off the midline, a useful imaging feature to differentiate them from primitive neuro-ectodermal tumours (PNETs),[1] hence the diagnostic radiological challenge posed by case 1. Both of our patients presented with supratentorial tumours, and our first patient's tumour was in the midline in a suprasellar location.
CT scanning usually demonstrates a hyperdense mass because of the high cellular density of teratoid/rhabdoid tumours.[2] MRI features of atypical teratoid/rhabdoid tumours include a bulky, heterogeneous mass with solid and cystic components, as described in our first patient. The heterogeneous nature of these tumours is due to the different cellular make-up associated with areas of necrosis, haemorrhage and calcifications.[4] Arslanoglu et al.[1] reported a 100% prevalence of calcification in histologically proven atypical teratoid/rhabdoid tumours. A peripheral rim of restricted diffusion on MRI, as described in both our patients, is an important imaging feature.[1, 2, 4] Low tumour ADC values compared with normal brain parenchyma are due to tumour hypercellularity.[1, 2, 4] Enhancement post contrast is variable in atypical teratoid/rhabdoid tumours. MRI spectroscopy is nonspecific as the findings are similar to PNET, showing elevated choline and low or absent N-acetyl aspartate.[4] Features that indicate aggressiveness and a poorer prognosis include obstructive hydrocephalus, as in our first patient, and cerebrospinal fluid seeding, as in our second patient,[4] which led to their death. The entire CNS must therefore be imaged at presentation to identify subarachnoid spread of the tumour. The differential diagnosis for atypical teratoid/rhabdoid tumour includes PNET, medulloblastoma, high-grade glioma and teratoma.[1, 3, 6]
The histopathological diagnosis of atypical teratoid/rhabdoid tumours is based on the presence of varying proportions of rhabdoid cells (which express vimentin, EMA and SMA) and primitive neuro-ectodermal cells. [1, 3, 4] Both cases described here demonstrated rhabdoid cells that were immunoreactive for vimentin and EMA. Classic rhabdoid phenotypic features may only be identified focally in an atypical teratoid/rhabdoid tumour. There is a broad spectrum of immunohistochemical expression, which is concordant with the histological diversity of the tumour. [3, 4, 6] Frequently, an atypical teratoid/rhabdoid tumour is confused with a PNET on histopathological evaluation.[1] However, specific immunohistological markers, in particular INI1, have made it possible to differentiate these tumours from PNETs in cases where primitive cells predominate.[2, 4]
Conclusion
Atypical teratoid/rhabdoid tumour of the CNS is a rare, highly aggressive childhood malignancy, and suprasellar location in particular is rare for this tumour. In the cases described here, CT and MRI demonstrated features of tumour calcification and peripheral restricted diffusion, which supported the diagnosis. Diagnosis requires biopsy and histopathological analysis using specific immunohistochemical markers. It is important for the radiologist to be aware of the imaging features of this rare tumour in patients with unusual presentations. When a calcified, heterogeneous mass with peripheral restricted diffusion and early CSF seeding is detected in a child younger than 2 years of age, the radiologist should consider the diagnosis of atypical teratoid/rhabdoid tumour.
References
1. Arslanoglu A, Aygun N, Tekhtani D, et al. Imaging findings of CNS atypical teratoid/rhabdoid tumors. AJNR Am J Neuroradiol 2004;25(3):476-480. [ Links ]
2. Bing F, Nugues F, Grand S, Bessou P, Salon C. Primary intracranial, extra-axial and supratentorial atypical rhabdoid tumor. Pediatr Neurol 2009;41(6):453-456. [http://dx.doi.org/10.1016/j.pediatrneurol.2009.07.019] [ Links ]
3. Reddy T. Atypical teratoid/rhabdoid tumor of the central nervous system. J Neurooncol 2005;75(3):309-313. [http://dx.doi.org/10.1007/s11060-005-6762-8] [ Links ]
4. Meyers S, Khademian Z, Biegel J, Chuang S, Korones D, Zimmerman R. Primary intracranial atypical teratoid/rhabdoid tumors of infancy and childhood: MRI features and patient outcomes. AJNR Am J Neuroradiol 2006;27(5):962-971. [ Links ]
5. Gandhi C, Krieger M, Mccomb J. Atypical teratoid/rhabdoid tumor: An unusual presentation. Neuroradiology 2004;46(10):834-837. [http://dx.doi.org/10.1007/s00234-004-1251-x] [ Links ]
6. Judkins AR, Eberhart CG, Wesseling P. Atypical teratoid/rhabdoid tumor. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, eds. WHO Classification of Tumours of the Central Nervous System. 4th ed. Lyon: IARC Press, 2007:147-149. [ Links ]
 Correspondence:
Correspondence:
N Mahomed
nasreen.mahomed@wits.ac.za

