










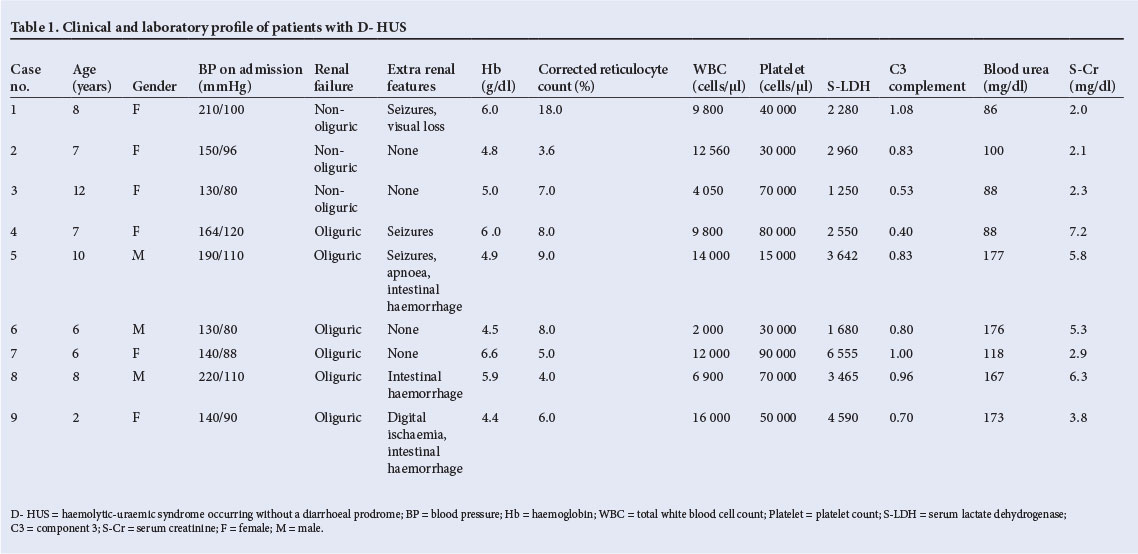
Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Child Health
On-line version ISSN 1999-7671
Print version ISSN 1994-3032
S. Afr. j. child health vol.8 n.2 Pretoria May. 2014
SHORT REPORT
Clinical profile and outcome of D- haemolytic-uraemic syndrome in children from south India
S UthupI; R C RadhakrishnanII
IMBBS, MD (Paediatrics), DM (Nephrology); Department of Paediatric Nephrology, SAT Hospital, Government Medical College, Thiruvananthapuram, Kerala, India
IIMBBS, MD (Paediatrics); Department of Paediatrics, SUT Academy of Medical Sciences, Thiruvananthapuram, Kerala, India
ABSTRACT
BACKGROUND: Haemolytic-uraemic syndrome (HUS) occurring without a diarrhoeal prodrome is termed D- HUS and has a poorer prognosis than D+ HUS, with high mortality and potential for long-term renal and non-renal morbidity.
METHODS: We studied nine children with D- HUS from the Pediatric Nephrology division of the Medical College, Trivandrum, India, over a period of 5 years. The clinical, haematological and renal profiles of all patients were recorded. All patients were treated with fresh-frozen plasma with or without dialysis.
RESULTS: The aetiology of HUS was not apparent in any of the cases, except in one patient who had a history of ayurvedic treatment for chronic cough. The mean age of the patients was 7.5 years with a male:female ratio of 1:2. Hypertension (HT) was present in eight cases (88%). Plasmapheresis was performed in 22% of patients; 67% underwent dialysis. Renal biopsy was performed in six patients. Four patients (44%) had good renal recovery while two progressed to end-stage renal disease. One child died in the acute phase, and one had a relapse of HUS. HT persisted in 44% and proteinuria in 33% of patients.
CONCLUSION: Early comprehensive management including dialysis, plasma infusions, and aggressive management of HT yields a good outcome in D- HUS. Persistent HT and progressive, chronic kidney disease require long-term management.
Haemolytic-uraemic syndrome (HUS) is an important cause of acute kidney injury (AKI) in children, leading to significant morbidity and mortality, and the effects of which may persist into adulthood.[1,2] HUS is characterised by a triad of micro-angiopathic haemolytic anaemia, thrombocytopenia and acute renal failure. HUS is usually characterised by oligoanuric renal failure similar to acute glomerulonephritis, but the outcome is guarded. Based on the presence or absence of a diarrhoeal prodrome, HUS may be classified as D+ or D-, respectively.
D- HUS or atypical HUS (aHUS) accounts for <10% of all HUS cases encountered. It is a heterogenous and sometimes familial disorder with complement dysregulation being the most important aetiological factor. The prognosis is worse, with death rates of up to 25% in the acute phase, and 50% of cases requiring ongoing renal replacement therapy.[3] The complement factor H (CFH), membrane cofactor protein (MCP) and complement factor I (CFI) genes are most commonly involved in familial HUS. Other factors predisposing to aHUS are infections (e.g. Streptococcus pneumoniae and HIV), drugs (e.g. mitomycin C, cisplatin, quinine, cyclosporin, tacrolimus and oral contraceptive pill), pregnancy and systemic diseases (systemic lupus erythaematosus, scleroderma, anti-phospholipid syndrome).[4]
Though rare in children, D- HUS is an important disorder in paediatric practice due to high mortality and potential for long-term renal and non-renal morbidity. The information on this disorder in children is very scant, especially from Asia. To enable clearer definition of the clinical characteristics of this syndrome, we studied D- HUS cases presenting to the paediatric nephrology division of a tertiary care teaching hospital in south India.
Methods
In this descriptive study we included patients who presented to the Pediatric Nephrology division of the Department of Paediatrics, Government Medical College, Thiruvananthapuram, Kerala, India, from 2007 to 2010. D- HUS was defined as evidence of AKI in association with micro-angiopathic haemolytic anaemia and thrombocytopenia (platelet count <100 000 cells/|µl) without a prodrome of diarrhoea. Children aged <12 years were included. The clinical and laboratory parameters at presentation and on follow-up were analysed. Hypertension (HT) was classified based on the 2004 Fourth report on high blood pressure in children and adolescents from the National High Blood Pressure Education Programme.[5] Good renal recovery was defined as a glomerular filtration rate (GFR) >80 ml/min on follow-up.
Results
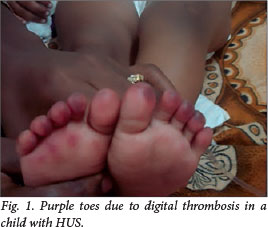
From September 2007 to September 2010, nine children (mean age 7.5 years; range 2 - 12) were admitted with D- HUS. The mean duration of follow-up was 52 months (range 38 - 66). The detailed clinical and laboratory profiles of all children are shown in Table 1. There was a female preponderance (male:female ratio 1:2). There were no identifiable risk factors in any patient except in one who gave a history of intake of ayurvedic medicines for chronic cough, the contents of which were unknown. At the time of admission, HT was present in 89%. Renal failure was oliguric in 67% and non-oliguric in 33%. Non-renal manifestations included seizures in three patients, apnoea in one, visual loss in one, left ventricular dysfunction (cardiomyopathy) in one and intestinal bleeding in three patients. One child with severe renal failure had purple discolouration of the toes, possibly due to thrombotic micro-angiopathy (Fig. 1). A Doppler study showed no evidence of deep-vein thrombosis (DVT) in the left lower limb. Major clinical features at the time of admission are shown in Table 2.
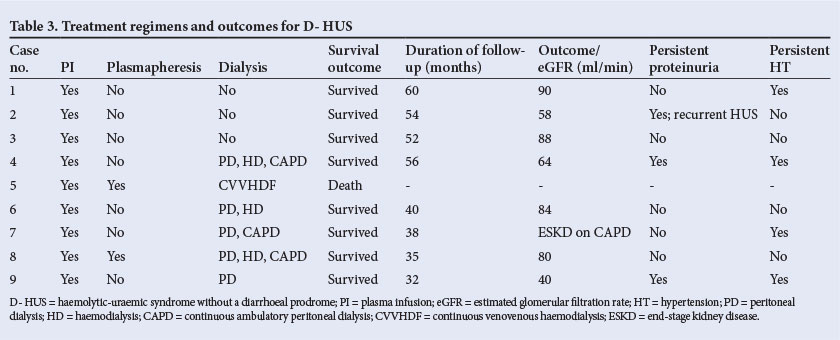
The average hospital stay was 45 days (range 14 - 120). Treatment regimens and outcomes are shown in Table 3. All patients received fresh plasma transfusions. Plasmapheresis was performed in two children. Six children (67%) had severe renal failure requiring dialysis. The mode of dialysis included acute peritoneal dialysis (PD) in two, acute PD followed by haemodialysis (HD) in three, and continuous venovenous HD (CVVHD) in one patient. Three children had severe, prolonged renal failure and were maintained on continuous ambulatory PD (CAPD); of these, two showed an improvement in renal function and were off dialysis after 6 months. One patient expired in the acute phase, despite plasmapheresis and CVVHD (11% mortality). He had severe involvement of the myocardium, intestinal bleeding and severe oligoanuric renal failure.
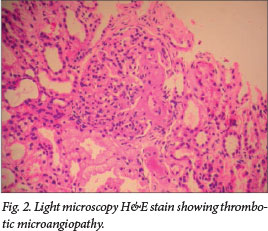
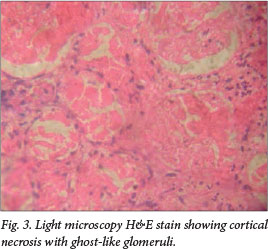
Low component 3 (C3) complement, indicating alternate complement pathway activation, was seen in 88%. Consent for renal biopsy was obtained in six patients, showing thrombotic microangiopathy in three (Fig. 2). Glomerular changes included focal segmental glomerulosclerosis (one patient), membrano-proliferative glomerulonephritis and focal proliferative glomerulonephritis (two patients each). One patient had severe cortical necrosis with ghost-like glomeruli and intra-arteriolar thrombosis (Fig. 3).
At follow-up, 44% had good renal recovery with a GFR >80 ml/min at 3 months. Two months after the onset of HUS, one child developed dilated cardiomyopathy with cardiac failure. She did not recover from kidney failure and is maintained on CAPD. Forty-four per cent of the series had persistent HT requiring the use of one or more anti-hypertensive drugs. The child with severe cortical necrosis had stage 3 chronic kidney disease (CKD) on follow-up. Recurrence of HUS was observed in one child, and was managed conservatively with plasma infusions (PIs), ACE inhibitors (ACEIs) and anti-hypertensives, with good renal recovery at 4 years of follow-up.
Discussion
HUS, first described by Gasser et al. in 1955,[6] is the most common cause of acute renal failure in children and is characterised by the triad of micro-angiopathic haemolytic anaemia, thrombocytopenia and acute renal failure. D+ HUS is the classic form, accounting for 95% of cases in children. It is preceded by a prodrome of diarrhoea, most commonly caused by an infection by shiga-toxin-producing Escherichia coli. D- HUS accounts for the remaining 5% of HUS cases and its aetiology, age at onset, and clinical presentations are far more varied. Unlike D+ HUS, D- HUS is not preceded by an identifiable gastrointestinal infection.
Clinically, D- HUS has been associated with various non-enteric bacterial and viral infections. Infection by S. pneumoniae has been linked to 40% of cases. Drugs, malignancies, transplantation, pregnancy, and other underlying medical conditions such as scleroderma and antiphospholipid syndrome have also been associated. Categories of drugs that have been most frequently associated with D- HUS include anti-cancer molecules (mitomycin, cisplatin, bleomycin, and gemcitabine), immunotherapeutics (cyclosporine, tacrolimus, OKT3, IFN and quinidine) and antiplatelet agents (ticlopidine and clopidogrel).
The pathogenesis of D- HUS has been the focus of current research and has, thus far, been associated with complement dysregulation in 30 - 50% of patients, with 14 - 33% having abnormalities in the CFH gene, 10 - 15% in the MCP gene and 2 - 13% in the CFI gene.[7-10] Mutations in these genes lead to unregulated amplification of the alternative pathway and deposition of activated complement on the surface of invading bacteria or damaged self-tissue, like apoptosed or inflamed renal endothelial cells.[11,12] The majority of these genes are situated in a cluster of complement regulatory genes on chromosome 1q32. The mutations are generally heterozygous, with patients having reduced (but not absent) activity of the factor, with autosomal dominant inheritance and 50% penetration. Homozygous and compound heterozygous mutations have also been described, usually with a more fulminant course.[13] The autosomal recessive ADAMTS13 deficiency, a familial form of the disease usually seen in children, is rare (2 - 3%). Acquired forms of ADAMTS13 deficiency, often associated with the presence of anti-ADAMTS13 antibodies, are more common in adults and older children.
Our study was aimed at describing the clinical profile and outcome of D- HUS. Unfortunately, mutational analyses to detect defects in complement regulatory genes could not be performed. The demographic profile of our series showed a greater incidence of D- HUS in girls, in concordance with the observations in diarrhoeal HUS.[14] Whether or not girls are more prone to develop HUS requires further investigation. Children with severe illness characterised by severe haemolysis (haemoglobin <5 g/dl, lactate dehydrogenase >2 500 IU), leukocytosis and severe renal failure at presentation, had a poorer outcome. Low C3 complement indicating alternate complement pathway activation was seen in 88% of the series. Forms of aHUS, especially those with mutations involving alternate complement pathway and decreased ADAMTS13 activity, are associated with signs of complement consumption. Thus, finding decreased C3 complement is a promising diagnostic parameter with good sensitivity for this form of HUS.[15]
Renal biopsy in our series showed thrombotic micro-angiopathy in three out of six patients. Fibrin thrombi in glomerular capillary loops are seen in glomerular HUS, while similar findings in arterioles suggest arteriolar HUS, which portends a poor prognosis. The evidence of thrombotic micro-angiopathy will be seen only in renal biopsies done in the acute phase of the illness. Glomerular changes that we encountered after the acute illness included focal segmental glomerulosclerosis, membranoproliferative glomerulonephritis and focal proliferative glomerulonephritis. These lesions indicate the response of glomeruli to ischaemic damage secondary to thrombotic occlusion of glomerular arterioles or capillaries. The progressive disease occurring in 40% of children with D- HUS is due to hyperfiltration damage occurring in surviving glomeruli and the impact of ongoing HT and proteinuria. The more frequent chronic renal lesion is characterised by the hyperfunction of nephrons remaining after the acute necrotising lesion, which leads to progressive scarring, and not by persistence or recurrence of the micro-angiopathic process.[16]
The patients in this series were managed by PIs with or without plasmapheresis and dialysis. Plasmatherapy has empirically become first-line treatment in aHUS, although no prospective, controlled trials have been conducted. Patients with mutations that induce complete or partial CFH quantitative deficiency may be controlled by PIs, but plasma exchanges appear to be more efficient in patients with mutations that result in a mutant dysfunctional CFH in the circulation[17] Early treatment is crucial.
There are scarce data about the long-term renal outcome following an episode of thrombotic micro-angiopathy. Three forms of progression to end-stage renal failure have been described. Children with most severe forms do not recover from acute renal failure and enter directly into a dialysis and transplantation programme. A second group recovers renal function partially, but continues to have persistent proteinuria and frequent HT, and progresses to end-stage renal failure in 2 - 5 years. In the third group, serum creatinine and creatinine clearance will become normal, but proteinuria persists; they are at risk of progressing to chronic renal failure and end-stage renal disease after >5 years, and sometimes as late as 20 years, after the acute disease.[16]
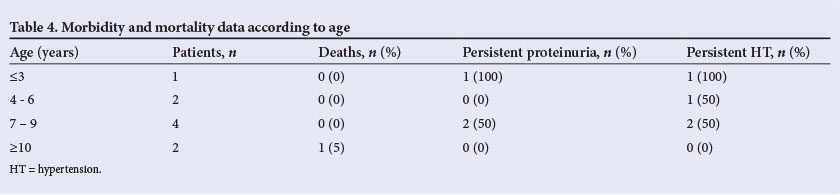
Reports suggest that up to 25% patients die in the acute phase, and 50% progress to end-stage renal failure in non-infection-related HUS.[8] The advent of plasma therapy has significantly improved the outcome of HUS due to ADAMTS13 deficiency, with mortality figures dropping from 80 - 90% to 10 - 20%.[18,19] In a large meta-analysis, it was found that death or end-stage renal disease occurs in about 12% of patients with diarrhoea-associated HUS, and 25% of survivors demonstrate long-term renal sequelae.[20] In a study on D- HUS from India, 38% of patients had relapses, 11% progressed to end-stage kidney disease, complete renal recovery was observed in 83% and 6% died.[21] In our study, good renal recovery, defined as a GFR >80 ml/min on follow-up, was found in 44% patients. Poor outcome, defined as a GFR <80 ml/min at 3 months, was seen in 44% with stage 5 CKD in one patient, stage 3 CKD in two patients and stage 2 CKD in one patient. The mortality in our series was 11%. One patient suffered multiple relapses and was managed by PIs during relapse. After the fourth relapse she was given prophylactic PIs monthly for 12 months. She did not have further relapses and is currently in stage 3 CKD on follow-up. Long-term, prophylactic plasmatherapy appears to be more efficient in preventing end-stage renal disease than plasmatherapy only during relapses.[17] We could not assess the prognostic implications of age on the final morbidity and mortality due to the small number of patients in our series, but there appears to be a trend towards persistent proteinuria in children aged <6 years (Table 4).
Conclusion
Prompt diagnosis and management of childhood D- HUS is a challenge for paediatricians and paediatric nephrologists alike. It is still difficult to understand the prognosis of this heterogeneous condition. It is highly possible that patients may still develop progressive renal disease after apparent renal recovery from D- HUS, especially with more children surviving severe disease. Hence, continued surveillance of these children is essential. There is evidence that early, comprehensive management improves the outcome and decreases the mortality and short-term morbidity; however, long-term follow-up and further studies with larger series are needed to understand this intriguing condition further, especially its management and prognostic determinants.
References
1. Westerholt S, Pieper A, Griebel M, et al. Characterization of the cytokine immune response in children who have experienced an episode of typical hemolytic-uremic syndrome. Clin Diagn Lab Immunol 2003;10(8):1090-1095. [ Links ]
2. Scheiring J, Andreoli SP, Zimmerhackl LB. Treatment and outcome of Shiga-toxin-associated hemolytic uremic syndrome [HUS]. Pediatr Nephrol 2008;23(10):1749-1760. [http://dx.doi.org/10.1007/s00467-008-0935-6] [ Links ]
3. Noris M, Remuzzi G. Non-shiga toxin-associated hemolytic uremic syndrome. In: Zipfel P, ed. Complement and Kidney Disease. Basel: Birkhauser-Verlag, 2005:65-83. [ Links ]
4. Kavanagh D, Goodship TH, Richards A. Atypical haemolytic uraemic syndrome. Br Med Bull 2006;77/78:5-22. [http://dx.doi.org/10.1093/bmb/ldl004] [ Links ]
5. National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents. Fourth report on high blood pressure in children and adolescents from National High Blood Pressure Education Programme. Paediatrics 2004;114(2 Suppl):555-576. [ Links ]
6. Siegler R, Oakes R. Hemolytic uremic syndrome; pathogenesis, treatment, and outcome. Curr Opin Pediatr 2005;17(2):200-204. [ Links ]
7. Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P. Genetics of HUS: The impact of MCP, CFH and IF mutations on clinical presentation, response to treatment, and outcome. Blood 2006;108:1267-1279. [http://dx.doi.org/10.1182/blood-2005-10-007252] [ Links ]
8. Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, Macher MA, Niaudet P, Guest G. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol 2007;18:2392-2400. [ Links ]
9. Kavanagh D, Richards A, Fremeaux-Bacchi V, et al. Screening for complement system abnormalities in patients with atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol 2007;2:591-596. [http://dx.doi.org/10.1681/ASN.2006080811] [ Links ]
10. Saunders RE, Goodship TH, Zipfel PF, Perkins SJ. An interactive web database of factor H-associated hemolytic uremic syndrome mutations: Insights into the structural consequences of disease associated mutations. Hum Mutat 2006;27:21-30. [http://dx.doi.org/10.1002/humu.20268] [ Links ]
11. Kavanagh D, Richards A, Atkinson J. Complement regulatory genes and hemolytic uremic syndromes. Annu Rev Med 2008;59:293-309. [http://dx.doi.org/10.1146/annurev.med.59.060106.185110] [ Links ]
12. Atkinson JP, Goodship TH. Complement factor H and the hemolytic uremic syndrome. J Exp Med 2007;204:1245-1248. [http://dx.doi.org/10.1084%2Fjem.20070664] [ Links ]
13. Cho HY, Lee BS, Moon KC, Ha IS, Cheong HI, Choi Y. Complete factor H deficiency-associated atypical hemolytic uremic syndrome in a neonate. Pediatr Nephrol 2007;22:874-880. [http://dx.doi.org/10.1007/s00467-007-0438-x] [ Links ]
14. Kinney JS, Gross TP, Porter CC, et al. Hemolytic-uremic syndrome: A population-based study in Washington, DC and Baltimore, Maryland. Am J Pub Health 1988;78(1):64-65. [http://dx.doi.org/10.2105/AJPH.78.L64] [ Links ]
15. Reusz GS, Szabo AJ, Reti M, et al. Diagnosis and classification of hemolytic uremic syndrome: The Hungarian experience. Transplant Proc 2011;43(4):1247-1249. [http://dx.doi.org/10.1016/j.transproceed.2011.03.071] [ Links ]
16. Repetto HA. Long-term course and mechanisms of progression of renal disease in hemolytic uremic syndrome. Kidney Int Suppl 2005;97:S102-S106. [http://dx.doi.org/10.1111/j.1523-1755.2005.09717.x] [ Links ]
17. Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T. Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost 2010;36(6):673-681. [ Links ]
18. Loirat C, Girma JP, Desconclois C, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura related to severe ADAMTS13 deficiency in children. Pediatr Nephrol 2009;24:19-29. [http://dx.doi.org/10.1007/s00467-008-0863-5] [ Links ]
19. Mannucci PM. Thrombotic thrombocytopenic purpura and the hemolytic uremic syndrome: Much progress and many remaining issues. Haematologica 2007;92:878-880. [ Links ]
20. Garg AX, Suri RS, Barrowman N, et al. Long-term renal prognosis of diarrhea-associated hemolytic uremic syndrome - a systematic review, meta-analysis, and meta-regression. JAMA 2003;290(10):1360-1370. [ Links ]
21. Shah N, Lobo V, Matnani M, Sharma J. Clinical Manifestations and Outcomes of D- [Atypical] Hemolytic Uremic Syndrome in Children. 20th National Conference Indian Society of Paediatric Nephrology, 2008. [ Links ]
 Correspondence:
Correspondence:
S Uthup
(susanuthup5@gmail.com)


{kind=link}
{kind=link}
{kind=link}