










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Child Health
On-line version ISSN 1999-7671
Print version ISSN 1994-3032
S. Afr. j. child health vol.8 n.1 Pretoria Feb. 2014
CASE REPORT
A case of incomplete and refractory Kawasaki disease: Diagnostic and therapeutic challenges
S MehndirattaI; K RajeshwariII; A P DubeyII; R KumarIII
IMBBS, DCh, DNB (Pediatrics), MNAMS; Department of Pediatrics, Lok Nayak Hospital, New Delhi, India
IIMD (Pediatrics); Department of Pediatrics, Maulana Azad Medical College, New Delhi, India
IIIMD PGT; Department of Pediatrics, Maulana Azad Medical College, New Delhi, India
ABSTRACT
The diagnosis of incomplete Kawasaki disease (KD) - i.e. cases that do not fulfil all the diagnostic criteria - requires a high index of suspicion. We report a case of incomplete KD that was refractory to treatment with intravenous immunoglobulin and subsequently responded to treatment with intravenous methylprednisolone.
Kawasaki disease (KD) is an acute febrile illness with vasculitis involving the medium-sized blood vessels. Its most serious effect is on the coronary arteries.[1-3] The incidence of KD appears to be rising on the Indian subcontinent, where it is increasingly being reported. This increase in reporting may be due to greater awareness of the disease.[4,,5] Not all cases fulfil all the diagnostic criteria, and these are termed incomplete KD.[6,7] We report a case of incomplete KD that was refractory to the recommended standard treatment with intravenous immunoglobulin (IVIG), but responded to treatment with intravenous methylprednisolone. The case highlights the diagnostic and therapeutic challenges posed by incomplete and refractory KD.
Case report
An 18-month-old boy was brought to hospital with a history of low-grade pyrexia for 5 days and respiratory distress for 2 days. On examination, he was febrile (axillary temperature 39.2°C), irritable and slightly pale. Examination of the respiratory system revealed a few crackles bilaterally. Initial routine laboratory investigations revealed a haemoglobin (Hb) concentration of 7.3 g/dl, a white cell count (WCC) of 23.4x109 cells/l and a platelet count of 53 9x109 cells/l. A provisional diagnosis of pneumonia was made and the patient was treated with intravenous antibiotics (ceftriaxone); however, the fever persisted and he remained irritable. The findings on cerebrospinal fluid examination were normal. After 3 days he developed a non-itchy erythematous rash all over his body. There was no history of joint pain or joint swelling. Urinary tract infection was excluded by routine urinalysis and culture. On day 9, slight swelling of the limbs developed and peeling at the tips of the fingers and toes was noted. The patient also had redness of the tongue and oral mucosa, but there was no conjunctival congestion and no signiicant lymphadenopathy. He responded only transiently to antipyretic therapy.
Repeat investigations revealed anaemia (Hb 5.6 g/dl), a high WCC (25.7x109 cells/l) with thrombocytosis (659x109 cells/l), a raised erythrocyte sedimentation rate (56 mm/h) and positive C-reactive protein (40 mg/l). Tests for rheumatoid factor and antinuclear antibodies were negative. Peripheral smear examination revealed dimorphic anaemia. The patient also had hyponatraemia (Na+ 128 mmol/l) and hypo-albuminaemia (albumin 25 g/l). Echocardiography revealed dilated ostia of the left main coronary artery (0.446 cm in diameter) (Fig. 1).
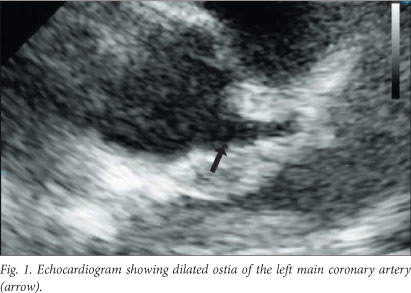
Owing to persistence of fever spikes up to 39.4°C, exclusion of other diagnoses and supportive laboratory findings, a diagnosis of incomplete KD was made. The patient was given IVIG (2 g/kg over 12 hours) and started on aspirin therapy (100 mg/kg/d in four divided doses). However, his fever persisted even after 48 hours, so IVIG was repeated. Once again there was no response. Because he was refractory to two doses of IVIG, the patient was given methylprednisolone as recommended (30 mg/kg pulse dose).[6] The pyrexia abated within 48 hours and the symptoms improved. He was discharged on treatment with aspirin (5 mg/kg/d). On follow-up about 6 months later, he was well, and echocardiography revealed that the coronary artery anomalies had resolved.
Discussion
The aetiology of KD is unclear. The clinical features, laboratory criteria and epidemiological proile strongly suggest an infectious cause, but despite a battery of investigations, results so far have been inconclusive. A typical case of KD can be diagnosed on the basis of a set of clinical and laboratory criteria and exclusion of other illnesses.[1-3] However, it is increasingly being recognised that not all children who subsequently develop complications fulil the criteria of typical KD. These cases are termed incomplete or atypical KD.[6,7] Approximately 10 - 15% of patients with KD fail to respond to treatment with IVIG, which is the standard irst-line treatment.
They have continuing vasculitis and require additional therapy, and are considered to have refractory KD.[8]
The exact incidence of incomplete KD is not known, with few cases being reported owing to the lack of diagnostic criteria and the presence of confounding clinical features that may mimic other childhood illnesses. These patients may remain undiagnosed, but they are as vulnerable to cardiac complications as patients with typical KD.[9] A diagnostic algorithm based on a combination of clinical and laboratory criteria has been proposed to help diagnose incomplete KD.[1] Factors associated with non-response to standard therapy have been described as male gender, hypo-albuminaemia, high aspartate aminotransferase levels and echocardiographic abnormalities at diagnosis.[10] Steroid therapy, e.g. intravenous methylprednisolone, is recommended for the treatment of refractory KD.[6,11,12] Complications of KD are coronary artery aneurysms, myocardial infarction, and sudden death, especially related to coronary artery involvement. However, studies have demonstrated that children who do not develop coronary anomalies may have prolonged endothelial dysfunction and abnormal lipid profiles on follow-up.[13] Newer therapeutic modalities that have been described, particularly for the treatment of refractory cases, are immunosuppressive agents (methotrexate, cyclophosphamide), infliximab (a chimeric murine/human immunoglobulin G-1 monoclonal antibody that binds specifically to human tumour necrosis factor-α), and plasma exchange. However, data on their use and success rates are limited.[14]
Our patient did not have lymphadenopathy or conjuctivitis, but persistent fever spikes, oedema of the hands and feet with skin peeling, oral mucosal changes and supportive laboratory findings helped to establish the diagnosis using the algorithm proposed for diagnosis of cases that do not fulfil all the criteria for KD.[1,6,7,15] Evidence of coronary ostia dilatation on the echocardiogram further supported the diagnosis. Other rheumatological illnesses such as systemic-onset juvenile rheumatoid arthritis (JRA) may have similar nonspecific clinical and laboratory features, and occasionally a case of evolving systemic JRA may mimic atypical KD.[16] Many clinical features of KD may mimic other childhood illnesses, and the diagnosis may be missed altogether or the case diagnosed late. The problem may be compounded by certain atypical presentations of KD such as associated hypertension, nephritis or seizures. However, awareness and the rate of diagnosis of the disease have increased in India over the past few years.[3-5]
In conclusion, the diagnosis and treatment of incomplete KD pose many challenges for the clinician. Since echocardiographic anomalies may not develop until 10 days into the illness, the decision to diagnose and treat early based only on clinical suspicion and nonspecific laboratory features may be problematic.
References
1. Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment and long-term management of Kawasaki disease: A statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young. Circulation 2004;110(17):2747-2771. [http://dx.doi.org/10.1161/01.CIR.0000145143.19711.78] [ Links ]
2. Rowley AH, Shulman ST. Pathogenesis and management of Kawasaki disease. Expert Rev Anti Infect Trier 2010;8(2):197-203. [http://dx.doi.org/10.1586/eri.09.109] [ Links ]
3. Consul M, Mishra S, Taneja A. Spectrum of Kawasaki disease. Indian J Pediatr 2011;78(4):488-490. [http://dx.doi.org/10.1007/s12098-010-0356-y] [ Links ]
4. Kushner HI, Macnee R, Burns JC. Impressions of Kawasaki syndrome in India. Indian Pediatr 2006;43(11):939-941. [ Links ]
5. Singh S, Aulakh R, Bhalla AK, et al. Is Kawasaki disease incidence rising in Chandigarh, North India? Arch Dis Child 2011;96(2):137-140. [http://dx.doi.org/10.1136/adc.2010.194001] [ Links ]
6. Parmar RC, Somale A, Bavdekar SB, Muranjan MN. Incomplete Kawasaki disease with recurrent skin peeling: A case report with the review of literature. J Postgrad Med 2003;49(1):72-74. [ Links ]
7. Witt M, Minich L, Bohnsack J, et al. Kawasaki disease: More patients are being diagnosed who do not meet American Heart Association criteria. Pediatrics 1999;104(1):e10. [http://dx.doi.org/10.1542/peds.104.Le10] [ Links ]
8. Freeman AF, Shulman ST. Refractory Kawasaki disease. Pediatr Infect Dis J 2004;23(5):463-464. [http://dx.doi.org/10.1097/01.inf.0000125893.66941.e0] [ Links ]
9. Baumer JH. Kawasaki disease: What to do with incomplete cases? Arch Dis Child Educ Pract Ed 2005;90(4):ep102-ep104. [http://dx.doi.org/10.1136/ adc.2005.088179] [ Links ]
10. Ashouri N, Takahashi M, Dorey F, Mason W. Risk factors for non response to therapy in Kawasaki disease. J Pediatr 2008;153(3):365-368. [http://dx.doi.org/10.1016/j.jpeds.2008.03.014] [ Links ]
11. Brogan PA, Bose A, Burgner D, et al. Kawasaki disease: An evidence based approach to diagnosis, treatment, and proposals for future research. Arch Dis Child 2002;86(4):286-290. [http://dx.doi.org/10.1136/adc.86.4.286] [ Links ]
12. Newburger JW, Fulton DR. Kawasaki disease. Current treatment Options in Cardiovascular Medicine 2007;9(2):148-158. [http://dx.doi.org/10.1007/s11936-007-0008-3] [ Links ]
13. McCrindle BW. Cardiovascular complications - coronary artery structure and function. Progress Pediatric Cardiol 2004;19(2):147-152. [http://dx.doi.org/10.1016/j.ppedcard.2004.08.008] [ Links ]
14. Rowley AH, Shulman ST. Recent advances in the understanding and management of Kawasaki disease. Curr Infect Dis Rep 2010;12(2):96-102. [http://dx.doi.org/10.1007/s11908-010-0091-6] [ Links ]
15. Kurutobi S, Nagai T, Kawakami N, Sano T. Coronary diameter in normal infants, children and patients with Kawasaki disease. Pediatr Int 2002;44(1):1-4. [http://dx.doi.org/10.1046/j.1442-200X.2002.01508.x] [ Links ]
16. Shaikh S, Ishaque S, Saleem T. Incomplete, atypical Kawasaki disease or evolving systemic juvenile idiopathic arthritis: A case report. Cases J 2009;2(1):6962. [http://dx.doi.org/10.4076/1757-1626-2-6962] [ Links ]
 Correspondence:
Correspondence:
S Mehndiratta
(drsmehndiratta@gmail.com)

