










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Child Health
On-line version ISSN 1999-7671
Print version ISSN 1994-3032
S. Afr. j. child health vol.8 n.1 Pretoria Feb. 2014
RESEARCH
Waardenburg syndrome in childhood deafness in Cameroon
J-J N NoubiapI; F DjomouI; R NjockI; G B ToureI; A WonkamII
IMD; Faculty of Medicine and Biomedical Sciences, University of Yaoundé, Yaoundé, Cameroon
IIMD; Division of Human Genetics, Department of Clinical Laboratory Sciences, Faculty of Health Sciences, University of Cape Town, South Africa
ABSTRACT
BACKGROUND: Waardenburg syndrome (WS) is a rare hereditary disorder essentially characterised by deafness and pigment disorders of the eyes, hair and skin.
METHODS: Between October 2010 and December 2011, we identified six patients with WS during an aetiological survey of 582 deaf participants recruited in schools for the deaf and ear, nose and throat outpatient clinics in seven of the ten regions of Cameroon. Two classic characteristics of WS were used as diagnostic criteria: deafness and pigmentation abnormalities (heterochromia iridis, white forelock and depigmented skin patches). In addition, to identify dystopia canthorum, a sign of WS type I, we calculated the W-index.
RESULTS: WS comprised 1% of the whole sample, 7% of the genetic cases, and 50% of the genetic syndromic cases. All patients with WS had severe to profound congenital sensorineural and symmetrical hearing loss with flat audiograms. They also had pigment disorders of the eyes and the skin. In the absence of dystopia canthorum, they were all classified as having WS type II. The pedigree was suggestive of autosomal dominant inheritance in two cases, and the four others seemed to be de novo cases.
CONCLUSION: The results suggest that WS type II is the most common syndromic form of hearing loss among Cameroonians. This has implications for retrospective genetic counselling and hearing tests for earlier management in affected families.
Waardenburg syndrome (WS) is an inherited disorder in which patients exhibit varying combinations of sensorineural hearing loss and abnormal pigmentation of the eyes, hair and skin.[1-4] WS is clinically heterogeneous and has been classified into four major types. Type I (WS1, MIM 193500) is characterised by deafness, dystopia canthorum, a broad nasal root, synophrys, hypoplasia of the alae nasi, and pigmentation abnormalities (heterochromia iridis, white forelock, depigmented skin patches);[3,4] type II (WS2, MIM 193510) presents like WS1, but without dystopia canthorum;[3,4] type III (WS3, MIM 148820), also known as Klein-Waardenburg syndrome, is an extreme presentation ofWS1, manifesting with upper-limb abnormalities (e.g. hypo-plasia, syndactyly)[3,4] and type IV (WS4, MIM 277580), also called Shah-Waardenburg syndrome or Waardenburg-Hirschsprung disease, combines pigmentation defects, deafness and Hirschsprung's disease.[3,4]
Types I and II are the most common, types IV and III being less frequent and very rare, respectively.[5] Overall it is estimated that between 1:20 000 and 1:40 000 people have WS, and there is a 1.43% prevalence among the congenitally deaf.[3,6]
Published data on deafness of genetic origin in sub-Saharan Africa are few and old. We recently carried out an aetiological survey on childhood deafness in Cameroon.[7] In the present article we report on the prevalence of WS and clinical and audiometric characteristics of the affected patients.
Methods
The study was approved by the National Ethics Committee of Cameroon (No. 123/CNE/SE/2010). Written informed consent was obtained from participants aged >18 years, and from parents or guardians of children aged <18 years, with assent from the child.
The study on causes of childhood hearing loss was carried out on 582 Cameroonians recruited in seven of the ten regions of Cameroon. Full details of clinical assessment and investigation procedures have been published elsewhere.[7]
Specifically, two classic characteristics of WS were used as diagnostic criteria: deafness and pigmentation abnormalities (heterochromia iridis, white forelock and depigmented skin patches). In addition, to identify dystopia canthorum, a sign of WS1, we calculated the W-index. Using a rigid ruler held against the face, we measured (in mm) the inner canthal distance (a), the interpupillary distance (b) and the outer canthal distance (c). We then calculated:
X = (2a - (0.2119c + 3.909))/c
Y = (2a - (0.2479b + 3.909))/b
W = X + Y + a/b
There is dystopia canthorum if the W-index is >1.95.[8]
Hearing levels were classified in accordance with recommendation number 02/1 of the Bureau International d'Audiophonologie, Belgium.[9]
Results
Frequency of WS
Of 582 patients with hearing loss, 86 (14.8%) had deafness of putative genetic origin, 12 cases being genetic syndromic deafness. We found six patients (three males and three females) with WS, representing 1% of the whole sample, 7% of genetic cases and 50% of genetic syndromic cases.
Clinical and audiological findings
Two of the patients with WS had a pedigree displaying autosomal dominant inheritance, each with an affected parent. The other patients had no close relatives with any features of WS, and hence appeared to be de novo cases. All the patients with WS were born to non-consanguineous parents.
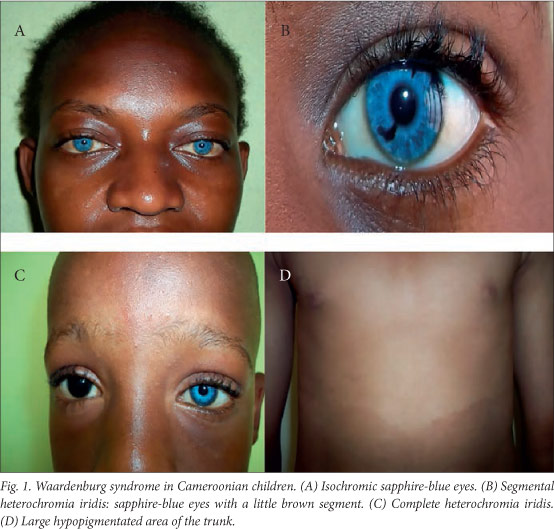
The clinical features are summarised in Table 1 and illustrated in Fig. 1. All the patients had normal psychomotor development, and none had Hirschsprung's disease or limb malformations. The W-index was <1.95 in all the patients, so in the absence of dystopia canthorum they were all classified as having WS2.
Management
Before enrolment, five of the six patients had never consulted an otorhinolaryngologist because of financial limitations and the absence of such a specialist in their towns; they had also not received prior genetic consultation. Furthermore, none had a hearing aid or cochlear implant. Five attended nursery schools or primary schools for the deaf, while one, aged 25 years, attended a secondary school for normal-hearing pupils despite his severe hearing loss.
Discussion
WS was the most frequent genetic syndrome found in our cohort, representing 50% of syndromic cases. This proportion is similar to the 62% prevalence of WS among black children with syndromic deafness in southern Africa found by Sellars et al.[10] WS could therefore be the most frequent form of syndromic deafness in sub-Saharan African populations. Sellars and Beighton[11] and Hageman[12] reported 3% and 1.7% prevalences of WS, respectively, among deaf pupils in southern Africa and Kenya - proportions higher than the 1% reported in this study. These data suggest that the prevalence of WS in childhood deafness may vary between 1% and 3% in sub-Saharan African populations.
We only found WS2 in our population. This finding differs from those of other studies in Africa. In their cohorts of patients with WS, Hageman[12] and de Saxe et al.[13] reported WS1:WS2 ratios of 18:12 and 31:21, respectively. Their reports suggest that WS1 may be more frequent than WS2. The absence of WS1 in our population seems to be a coincidental finding that could be related to poor availability of, and access to medical services in some areas of the country. In addition, for financial reasons many children with hearing loss may not have access to schools for the deaf, and have to choose between attending a normal school and not attending school at all. We did not find any cases of WS3 and WS4, which are rare in Caucasians.[5] The lone report we found of WS4 in Africa was a case in a newborn from Morocco.[14] Hirschsprung's disease, a major feature in WS4, accounts for a significant part of the early childhood mortality in these patients. In Africa, some patients with WS4 may die before diagnosis because of inadequate access to healthcare.
WS has a very high phenotypic variability.[3] Apart from one patient with premature canitis, none of our patients exhibited depigmentation of the hair, which has been seen in Nigerian children with WS, presenting as white forelock as in Caucasians.[3,15] All of our patients had hypoplastic blue eyes, and two of the six had partial or complete heterochromia of the iris, which occurs in 21 - 28% of patients with WS.[16]
WS is mainly transmitted as an autosomal dominant trait, but can also result from recessive inheritance or de novo occurrence.[3,6]-Because of the variable phenotype and incomplete penetrance,[3] close relatives of affected patients who exhibit very slight features may be undiagnosed. Sporadic cases can therefore be de novo, autosomal dominant or recessive.
None of our patients had prior genetic consultation. A medical genetic service was recently inaugurated in Cameroon,[17] so genetic counselling and molecular studies for deafness will become more possible in the country. Given that our patients have WS2, they may have mutations on the MTIF, SOX10 and SNAI2 genes.[6] However, molecular studies can be ineffective, as 85% of WS2 cases are unexplained at the molecular level.[18]
Except for Hirschsprung's disease in WS4, hearing loss is the most important factor in all WS types, because of the impairment of quality of life. As congenital deafness is generally diagnosed late in sub-Saharan African countries,[7] iris hypoplasia and heterochromia iridis are the main clinical features that could enable early diagnosis of WS and subsequent investigation for hearing loss. The best management for sensorineural hearing loss in childhood is cochlear implantation;[19] however, this approach is unfortunately not available in Cameroon.
Conclusion
The results suggest that WS2 is the most common syndromic form of hearing loss among Cameroonians. This has implications for retrospective genetic counselling and hearing tests for earlier management in affected families.
Author contributions
JJNN contributed to the study design, collected the data and drafted the manuscript. FD, RN and GBT contributed to the study design and revised the manuscript. AW conceived the study, supervised the research and drafted the manuscript. All authors approved the final version of the manuscript.
Acknowledgements
We are grateful to the patients and their parents for their participation in this research.
References
1. Hageman MJ, Delleman JW. Heterogeneity in Waardenburg syndrome. Am J Hum Genet 1977;29(5):468-485. [ Links ]
2. Markova TG, Megrelishvilli SM, Shevtsov SP, Shvarts EI. Clinical and molecular genetic investigation of Waardenburg syndrome type 1. Vestn Otorinolaringol 2003;(1):17-19. [ Links ]
3. Read AP, Newton VE. Waardenburg syndrome. J Med Genet 1997;34(8):656-665. [http://dx.doi.org/10.1136/jmg.34.8.6561 [ Links ]
4. Newton VE. Clinical features of the Waardenburg syndromes. Adv Otorhinolaryngol 2002;61:201-208. [http://dx.doi.org/10.1159/0000668101 [ Links ]
5. Haddad NM, Ente D, Chouery E, et al. Molecular study of three Lebanese and Syrian patients with Waardenburg syndrome and report of novel mutations in the EDNRB and MITF genes. Mol Syndromol 2011;1(4):169-175. [http://dx.doi.org/10.1159/000322891] [ Links ]
6. Pingault V, Ente D, Dastot-Le Moal F, et al. Review and update of mutations causing Waardenburg syndrome. Hum Mutat 2010;31(4):391-406. [http:// dx.doi.org/10.1002/humu.21211] [ Links ]
7. Wonkam A, Noubiap JJ, Djomou F, Fieggen K, Njock R, Toure GB. Etiology of childhood hearing loss in Cameroon (sub-Saharan Africa). Eur J Med Genet 2013;56(1):20-25. [http://dx.doi.org/10.1016/j.ejmg.2012.09.010] [ Links ]
8. Newton VE. Waardenburg's syndrome: A comparison of biometric indices used to diagnose lateral displacement of the inner canthi. Scand Audiol 1989;18(4):221-223. [http://dx.doi.org/10.3109/01050398909042198] [ Links ]
9. Bureau International d'Audiophonologie. Classification audiométrique des déficiences auditives. 1997. http://www.biap.org/recom02-1.htm (accessed 21 March 2010). [ Links ]
10. Sellars S, Beighton G, Horan F, Beighton P. Deafness in black children in southern Africa. S Afr Med J 1977;51(10):309-312. [ Links ]
11. Sellars S, Beighton P. The Waardenburg syndrome in deaf children in southern Africa. S Afr Med J 1983;63(19):725-728. [ Links ]
12. Hageman MJ. Waardenburg's syndrome in Kenyan Africans. Trop Geogr Med 1978;30(1):45-55. [ Links ]
13. De Saxe M, Kromberg JG, Jenkins T. Waardenburg syndrome in South Africa: Part I. An evaluation of clinical indings in 11 families. S Afr Med J 1984;66(7):256-261. [ Links ]
14. Mahmoudi A, Rami M, Khattala K, Elmadi A, Afifi MA, Youssef B. Shah-Waardenburg syndrome. Pan Afr Med J 2013;14:60. [http://dx.doi.org/10.11604/pamj.2013.14.60.1543] [ Links ]
15. Abah ER, Oladigbolu KK, Samaila E, Merali H, Ahmed AO, Abubakar TH. Ophthalmologic abnormalities among deaf students in Kaduna, Northern Nigeria. Ann Afr Med 2011;10(1):29-33. [http://dx.doi.org/10.4103/1596-3519.76573] [ Links ]
16. Tagra S, Talwar AK, Walia RL, Sidhu P. Waardenburg syndrome. Indian J Dermatol Venereol Leprol 2006;72(4):326. [http://dx.doi.org/10.4103/0378-6323.26718] [ Links ]
17. Wonkam A, Ngongang Tekendo C, Sama DJ, et al. Initiation of a medical genetics service in sub-Saharan Africa: Experience of prenatal diagnosis in Cameroon. Eur J Med Genet 2011;54(4):e399-e404. [http://dx.doi.org/10.1016/j.ejmg.2011.03.013] [ Links ]
18. Bondurand N, Dastot-Le Moal F, Stanchina L, et al. Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am J Hum Genet 2007;81(6):1169-1185. [http://dx.doi.org/10.1086/522090] [ Links ]
19. De Sousa Andrade SM, Monteiro AR, Martins JH, et al. Cochlear implant rehabilitation outcomes in Waardenburg syndrome children. Int J Pediatr Otorhinolaryngol 2012;76(9):1375-1378. [http://dx.doi.org/10.1016/j.ijporl.2012.06.010] [ Links ]
 Correspondence:
Correspondence:
A Wonkam
(ambroise.wonkam@uct.ac.za)


{kind=link}