










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Child Health
versão On-line ISSN 1999-7671
versão impressa ISSN 1994-3032
S. Afr. j. child health vol.7 no.4 Pretoria Abr. 2013
SHORT REPORT
Pulmonary alveolar proteinosis in a child from an informal settlement: 12 litres of fluid drained from the lungs and successful use of ECMO
D A WhiteI; S R KlugmanII; R WeilIII; E ZigiriadisIV; R J GreenV
IMB BCh, FCPaed (SA), MMed (Paed), Dip Allerg (SA), Cert Pulm SA (Paed); Division of Paediatric Pulmonology, Charlotte Maxeke Johannesburg Academic Hospital and Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIMB BCh, FCPaed (SA); Division of Paediatric Pulmonology, Charlotte Maxeke Johannesburg Academic Hospital and Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIIMB BCh, DCH, Dip Allerg (SA); Division of Paediatric Pulmonology, Charlotte Maxeke Johannesburg Academic Hospital and Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IVMB BCh, FC Cardio (SA); Division of Cardiothoracic Surgery, Charlotte Maxeke Johannesburg Academic Hospital and Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
VMB BCh, FCPaed (SA), PhD; Division of Paediatric Pulmonology, Steve Biko Academic Hospital and Faculty of Health Sciences, University of Pretoria, Pretoria, South Africa
Pulmonary alveolar proteinosis (PAP) is a rare cause of chronic interstitial lung disease, characterised by accumulation of pulmonary surfactant, respiratory insufficiency and an increased incidence of infections. The current standard therapy is whole-lung lavage to remove the accumulated surfactant. We report on a cachexic 12-year-old boy from an informal settlement in South Africa, presenting for the first time with PAP. Twelve litres of broncho-alveolar lavage fluid were drained under extracorporeal membrane oxygenation, and the patient gained 10 kg during his 2-month admission.
Pulmonary alveolar proteinosis (PAP) is a syndrome characterised by respiratory failure caused by pulmonary surfactant accumulation and resulting in respiratory insuiciency and an increased incidence of infections.[1] The current standard therapy is whole-lung lavage, which is used to physically remove the accumulated surfactant.[2] PAP can be grouped into distinct categories based on clinical, histopathological, biochemical and genetic data.[3]
Surfactant homeostasis is critical for lung function and is tightly regulated, in part by pulmonary granulocyte-macrophage colony-stimulating factor (GM-CSF), which is required for surfactant clearance by alveolar macrophages and alveolar macrophage maturation[4] The effects of GM-CSF are mediated by cell-surface receptors composed of GM-CSF-binding a-chains and affinity-enhancing β-chains (encoded by CSF2RA and CSF2RB, respectively)[4] Primary PAP occurs when GM-CSF signalling is disrupted[3] either on an auto-immune basis, where high levels of neutralising GM-CSF auto-antibodies block GM-CSF signalling (90% of cases), or by GM-CSF receptor dysfunction. Interestingly, recent articles describing both CSF2RA mutations[2,3] and CSF2RB mutations[4] in children with PAP failed to detect GM-CSF auto-antibodies in any of the patients, and identified increased serum GM-CSF as a useful biomarker. Secondary PAP occurs as a consequence of an underlying disease presumed to impair surfactant clearance by reducing either the numbers or the functions of alveolar macrophages[3] Hereditary disorders of surfactant production, for example, because of mutations in the genes encoding surfactant protein (SP)-B, SP-C or ABCA3, exhibit disordered surfactant homeostasis to varying degrees but are distinguished from PAP by their surfactant dysfunction, disruption of alveolar wall architecture, and clinical course.[3]
Case report
A thin 12-year-old boy on a stretcher was wheeled into the pulmonology clinic at Charlotte Maxeke Johannesburg Academic Hospital in Gauteng Province, South Africa. He had been admitted to a local district hospital a month before after presenting with a short history of chest pain and dyspnoea. He had received repeated courses of antibiotics for pneumonia, but remained oxygen dependent. His cardiac status was normal and he was HIV-uninfected. His symptoms had not responded to 3 months of empiric antituberculosis therapy. He was born and had lived for most of his life in a rural part of KwaZulu-Natal Province and had no unusual environmental exposures. He had recently moved to Gauteng and was living with his mother in an informal settlement.
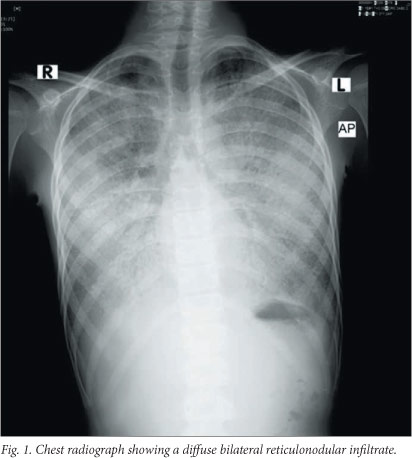
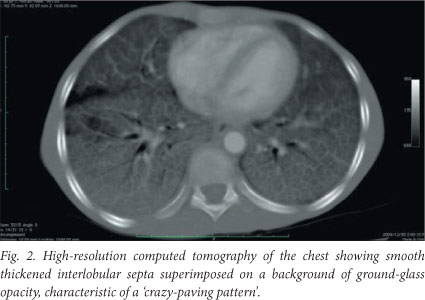
On examination the patient was cachexic, weighing only 20 kg (50% of expected weight for age). He was alert, and on oxygen via face mask with an oxygen saturation of 92%. He became severely cyanosed off oxygen. He had digital clubbing and no significant lymphadenopathy. He was tachypnoeic with fine inspiratory crackles at both lung bases. There were no signs of cor pulmonale. Pulmonary function testing revealed severe restrictive impairment. A chest radiograph revealed a diffuse bilateral reticulonodular infiltrate (Fig. 1), and high-resolution computed tomography (HRCT) of the chest showed smooth thickened interlobular septa superimposed on a background of ground-glass opacity, characteristic of a 'crazy-paving pattern' (Fig. 2) and in keeping with an interstitial lung disease, most likely PAP. Haematological testing revealed a haemoglobin concentration of 18 g/dl, but levels of electrolytes, C-reactive protein, a beta-D-glucan assay, an autoimmune screen and immunoglobulins and their subclasses were all normal. Smear and culture of the sputum for tuberculosis (TB) was negative. A barium swallow examination revealed mild gastro-oesophageal reflux which was treated with omeprazole.
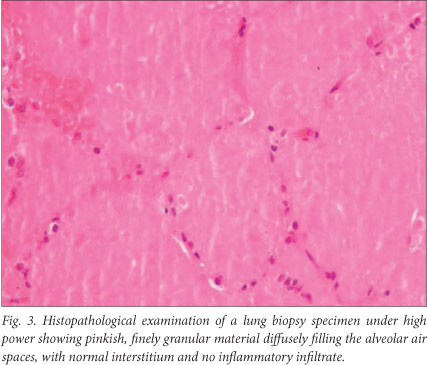
The findings on bronchoscopy were normal, and broncho-alveolar lavage (BAL) only produced a small amount of milky secretions. An open biopsy of the left lung was complicated by a pneumothorax, which required two intercostal drains. Postoperatively the patient required mechanical ventilation in the intensive care unit. The lung biopsy revealed diffuse filling of the alveoli by a pinkish, granular exudate that stained periodic acid-Schiff (PAS) positive and normal pleura and interstitium, diagnostic of PAP (Fig. 3).
Numerous subsequent attempts at BAL using a double-lumen bronchoscope and saline washes of both lungs under conventional ventilation failed, as the patient sustained cardiorespiratory arrests secondary to hypoxia and the procedures had to be aborted. He was given a single dose of GM-CSF subcutaneously but remained ventilator dependent. As there was no evidence of TB, the TB treatment was stopped. Finally, with the patient on extracorporeal membrane oxygenation (ECMO) in theatre, 12 litres of milky, pink fluid was successfully drained out of both lungs (Fig. 4).

The patient improved dramatically postoperatively; his pneumothorax re-expanded, and he was extubated successfully. His oxygen requirements decreased and he eventually only needed nasal prong oxygen at night. Careful attention to nutrition with supplements enabled him to gain 10 kg during his admission.
We then faced another obstacle. The patient's mother lived in an informal settlement under very poor social circumstances with no electricity, and therefore did not qualify for home oxygen. We placed him in a children's home with oxygen at night and enrolled him in our hospital school. He was discharged after a 2-month hospital stay and is currently doing very well.
Discussion
The diagnosis of PAP can be made with confidence on the appearance of the lung on HRCT of the thorax, which shows patchy or diffuse ground-glass opacifications with superimposed interlobular and intralobular septal thickening, a pattern referred to as 'crazy-paving',[5] in conjunction with examination of fluid obtained from segmental alveolar lavage.[6] The lavage fluid has an opaque milky appearance. [7] It contains foamy alveolar macrophages and increased numbers of lymphocytes, but few inflammatory cells of other types.[7] There are also large, acellular, eosinophilic bodies in a difuse background of granular material that stains with PAS, as well as elevated levels of surfactant proteins.[7] Lung biopsy is not always required, and in view of the patchy nature of the disease may be inconclusive.[6] Tests for GM-CSF auto-antibodies and genetic testing for GM-CSF receptor abnormalities are not currently available in South Africa.
Treatment of PAP depends on the underlying cause. Autoimmune PAP has been treated successfully by whole-lung lavage, which improves clinical, physiological and radiographic findings as well as survival.[7] The median duration of clinical benefit from lavage has been reported to be 15 months.[7] Genetic/hereditary PAP (GM-CSF receptor dysfunction) in paediatric patients has responded well to whole-lung lavage, similar to results for patients with autoimmune PAP and in contrast to patients with disorders of surfactant production, who do not respond well.[2] Multiple segmental lavage by fibre-optic bronchoscopy is a possible alternative to whole-lung lavage and has been used successfully in infants, especially as a feasible alternative in resource-constrained environments.[8] Therapy for secondary PAP involves treating the underlying condition.[7] Administration of GM-CSF subcutaneously to adult patients with idiopathic PAP has resulted in a partial favourable response, manifest by a decreased need for oxygen.[9] Newer potential therapies include aerosolised GM-CSF therapy, gene therapy for GM-CSF receptor defects, and bone marrow transplantation.[3]
Despite a huge burden of infectious diseases in South Africa, rare lung diseases still occur. In addition, despite a general paucity of social services, some children still receive health services comparable to the best in the world.
Acknowledgements. We thank Professor Guy Richards for his contribution to the patient's intensive care, the rest of the cardiothoracic surgical team for their contributions to his care, and the nursing staff in the intensive care unit and paediatric ward at Charlotte Maxeke Johannesburg Academic Hospital.
References
1. Trapnell BC, Carey BC, Uchida K, Suzuki T. Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Curr Opin Immunol 2009;21(5):514-521. [http://dx.doi.org/10.1016%2Fj.coi.2009.09.004] [ Links ]
2. Suzuki T, Sakagami T, Young L, et al. Hereditary pulmonary alveolar proteinosis: Pathogenesis, presentation, diagnosis and therapy. Am J Respir Crit Care Med 2010;182(10):1292-1304. [http://dx.doi.org/10.1164%2Frccm.201002-0271OC] [ Links ]
3. Suzuki T, Sakagami T, Rubin BK, et al. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J Exp Med 2008;205(12):2703-2710. [http://dx.doi.org/10.1084%2Fjem.20080990] [ Links ]
4. Suzuki T, Maranda B, Sakagami T, et al. Hereditary pulmonary alveolar proteinosis caused by recessive CSF2RB mutations. Eur Respir J 2011;37(1):201-217. [http://dx.doi.org/10.1183%2F09031936.00090610] [ Links ]
5. De Blic J. Pulmonary alveolar proteinosis in children. Paediatr Respir Rev 2004;5(4):316-322. [http://dx.doi.org/10.1016%2Fj.prrv.2004.07.001] [ Links ]
6. Shah PL, Hansell D, Lawson PR, Reid KBM, Morgan C. Pulmonary alveolar proteinosis: Clinical aspects and current concepts on pathogenesis. Thorax 2000;55(1):67-77. [http://dx.doi.org/10.1136%2Fthorax.55.L67] [ Links ]
7. Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med 2003;349(26):2527-2539. [http://dx.doi.org/10.1056%2FNEJMra023226] [ Links ]
8. Hodges O, Zar HJ, Mamathuba R, Thomas J. Bilateral partial lung lavage in an infant with pulmonary alveolar proteinosis. Br J Anaesth 2010;104(2):228-230. [http://dx.doi.org/10.1093%2Fbja%2Faep371] [ Links ]
9. Seymour JF, Presneill JJ, Schoch OD, et al. Therapeutic efficacy of granulocyte-macrophage colony-stimulating factor in patients with idiopathic acquired alveolar proteinosis. Am J Respir Crit Care Med 2001;163(2):524-531. [http://dx.doi.org/10.1164%2Fajrccm.163.2.2003146] [ Links ]
 Correspondence: R J Green (robin.green@up.ac.za)
Correspondence: R J Green (robin.green@up.ac.za)

