










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Child Health
On-line version ISSN 1999-7671
Print version ISSN 1994-3032
S. Afr. j. child health vol.7 n.2 Pretoria May. 2013
CASE REPORT
Compound heterozygous sickle cell disease and β0-thalassaemia: An interesting case
R von FintelI; R SchwyzerII; J PooleIII; N A AlliIV
IMB BCh, FCPaed (SA). Department of Paediatric Haematology/Oncology, Charlotte Maxeke Academic Hospital and Wits Donald Gordon Medical Centre, Johannesburg, South Africa
IIMB BCh, DCH. Department of Paediatric Haematology/Oncology, Charlotte Maxeke Academic Hospital and Wits Donald Gordon Medical Centre, Johannesburg, South Africa
IIIMB BCh, DCH, FCP. Department of Paediatric Haematology/Oncology, Charlotte Maxeke Academic Hospital and Wits Donald Gordon Medical Centre, Johannesburg, South Africa
IVMB BCh, FCPath (Haem) (SA). Department of Haematology, School of Pathology, National Health Laboratory Service, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
Haemoglobinopathies are a group of inherited disorders caused by structural variations of the haemoglobin molecule. We report the case of a 5-year-old girl suffering from chronic haemolytic anaemia. A diagnosis of compound heterozygous sickle cell disease (SCD) and β0-thalassaemia was established using haemoglobin sub-fractionation studies and parental screening. The latter revealed the father and mother to have heterozygous SCD and heterozygous β-thalassaemia, respectively. Evaluation of children with a haemolytic anaemia requires a thorough history and physical examination, appropriate laboratory testing and careful interpretation of the results.
Case report
A 5-year-old girl was referred from a regional hospital to the paediatric haematology clinic at Charlotte Maxeke Johannesburg Academic Hospital, Johannesburg, South Africa, with severe anaemia. She presented with an acute history of vomiting and abdominal pain.
Past history. The patient had been born at term by normal vaginal delivery. On day 3 of life she was noted to have jaundice and required phototherapy for 3 days. Subsequently she had been well, with no further significant medical history or hospital admissions. Both parents were of coloured descent and were unaware of their carrier status. The father was from Mitchell's Plain near Cape Town, and the mother was from Johannesburg. Although the mother's distant family was of Asian descent, there was no family history of a haemoglobinopathy.
Clinical examination. The child's weight and height were 14.9 kg (3rd percentile) and 109.5 cm (50th percentile), respectively. Her head circumference was 49 cm. She was markedly pale and also had mild clinical jaundice. There was no significant lymphadenopathy. Signs of extramedullary erythropoiesis were present, notably frontal bossing and maxillary hyperplasia (Fig. 1) and an 8 cm splenomegaly. Cardiovascular examination revealed mild tachycardia, but there were no features of congestive cardiac failure. The liver was palpable 2 cm below the right costal margin, and the rest of the systemic examination was unremarkable.
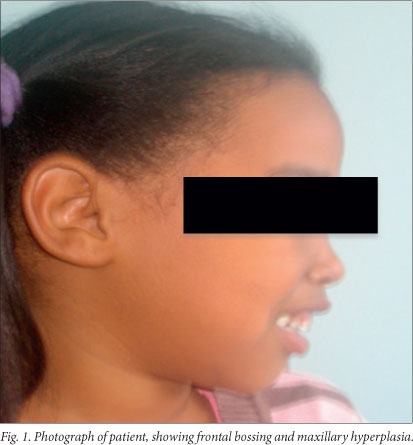
The patient had a 3-year-old sibling who was asymptomatic. She was brought to our clinic on a subsequent visit, where she was noted to have a 2 cm splenomegaly but no overt maxillary hyperplasia.
Methods and results
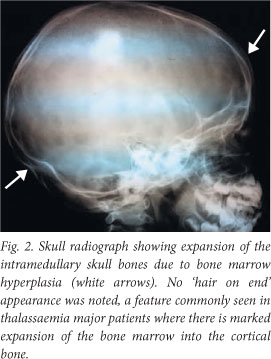
A skull radiograph showed expansion of intramedullary skull bones due to bone marrow hyperplasia (Fig. 2).
The results of laboratory investigations are summarised in Table 1. A full blood count revealed a normocytic, hypochromic anaemia with a mild thrombocytopenia and leuko-erythroblastic reaction (left shift and presence of nucleated red cells). There was evidence of ongoing haemolysis, viz. a decreased haptoglobin level, unconjugated hyberbilirubinaemia, elevated serum lactate dehydrogenase and reticulocytosis.
The peripheral blood smear showed numerous sickle cells, confirming the presence of sickle cell disease (SCD). However, other red cell features noted were suggestive of a possible additional thalassaemic component, viz. target cells and 'folded' red cells (Fig. 3).
Haemoglobin (Hb) separation studies were done on the patient as well as other family members, viz. both parents and sister, to investigate a suspected haemoglobinopathy. This included high- performance liquid chromatography (HPLC) and Hb electrophoresis. In both methods, the charge difference between Hb subtypes is exploited for their separation.[1]
It should be noted that for Hb sub-fractionation, the use of two different methods is recommended since no single method can separate all Hb subtypes. The Bio-Rad D10 instrument was used for HPLC analysis. Hb electrophoresis was performed on the Sebia instrument, which separates the Hb subtypes on a mylar-backed gel membrane. The separated bands are visualised after staining with a general protein stain.[1]
Interpretation of results
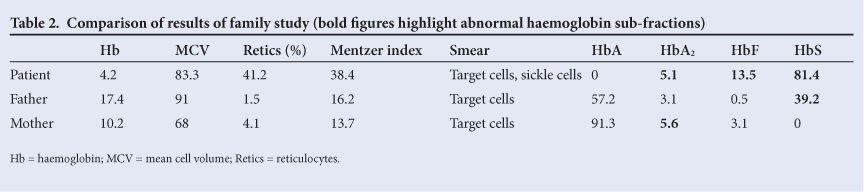
Hb sub-fractionation and blood count results for the patient and both parents are set out in Table 2. Based on the above investigations, the following diagnostic conclusions were reached:
1. The father had heterozygous SCD, since both HbS and HbA were present.
2. The mother was heterozygous for β-thalassaemia, as evidenced by the microcytosis and elevated HbA2 level.
3. The patient had compound heterozygous SCD and β0-thalassaemia (SCD/β0thal). In view of the absence of HbA, it could be concluded that the thalassaemia was of the β0 variety, i.e. there is no output of β-globin chains from the affected β-globin gene.
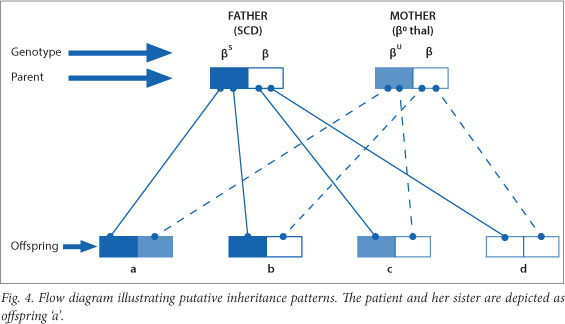
The patient's younger sister was also tested, and the results indicated that she too had SCD/β0thal. This is unfortunate, as the probability of inheriting both the βS- and β0-thalassaemia genes from the father and mother respectively is 25%.
The various combinations of inheritance are depicted as a flow diagram in Fig. 4. Each combination has a 25% probability of transmittance.
Discussion
Inherited Hb disorders constitute the most common single-gene defects in man.[2] The mode of inheritance is autosomal recessive.
They are classified into two main groups:
1. The thalassaemias, caused by decreased or absent production of normal globin chains.[3]
2. Hb variants, caused by production of abnormal globin chains (e.g. HbS, HbC).[3]
The patient described here had a β0-thalassaemia mutation in one β-globin gene and a βS mutation in the allelic β-globin gene. HbS constitutes the major Hb sub-fraction, since output of normal β-globin chains is zero. Although the clinical course of SCD/β0thal is generally less severe than that of homozygous SCD (SSD), with lower mortality rates,[4,5] patients are rarely asymptomatic. The lower morbidity and mortality rates can be explained by relatively lower HbS levels that result from higher HbA2 and HbF levels. Patients with SCD/ β0thal with unusually high levels of HbF have a mild phenotype, as was the case with our patient, who had an HbF level of 15%. HbF and HbA2 both have inhibitory effects on the sickling process as they do not participate in the formation and elongation of sickle tactoids. However, despite the absence of acute sickle crises there has been ongoing, albeit compensated, haemolysis with clinical consequences of long-term haemolysis.
The cause of the abdominal pain and vomiting appears to be infective (high white cell count, elevated C-reactive protein), but an acute abdominal crisis with or without an infective element cannot be ruled out. It should be noted that infection is a common precipitant of an acute sickle crisis.
Acute splenic sequestration crisis is a life-threatening complication in SSD, where the spleen enlarges rapidly. As a consequence, there is sudden pooling of blood in the spleen that leads to a reciprocal drop in the Hb. This risk diminishes in late childhood as the spleen progressively atrophies to a fibrotic mass. The incidence of splenic sequestration crisis in SCD/β0-thal is higher than in SSD, since the spleen is largely preserved and often enlarged in SCD/β0-thal.[6] For this reason the increased risk of splenic sequestration crisis in SCD/β0-thal continues into adulthood. Our patient had a fairly large spleen in the steady state, and there was no clinical evidence of further enlargement to suspect splenic sequestration crisis. The moderately severe anaemia can be attributed to a combination of factors including hypersplenism, reticulo-endothelial iron blockade due to infection, and possible abdominal crisis.
Co-inheritance of βS- and β-thalassaemia mutations is unusual in people of South African coloured descent. However, there is a high probability of crossover from the South African Malay gene pool, which has its origins in Malaysia. According to the separatist policies of the previous political dispensation (prior to 1994), Malays and coloureds were regarded as one ethnic group and were accordingly assigned the same residential areas and amenities.
This case underlines the importance of a thorough history, clinical examination and appropriate laboratory testing for suitable management and prognostication as well as genetic counselling.[7] Parental studies proved crucial in confirming the double heterozygous state of SCD/β0-thal, especially since resources for genetic studies in our setting are limited.
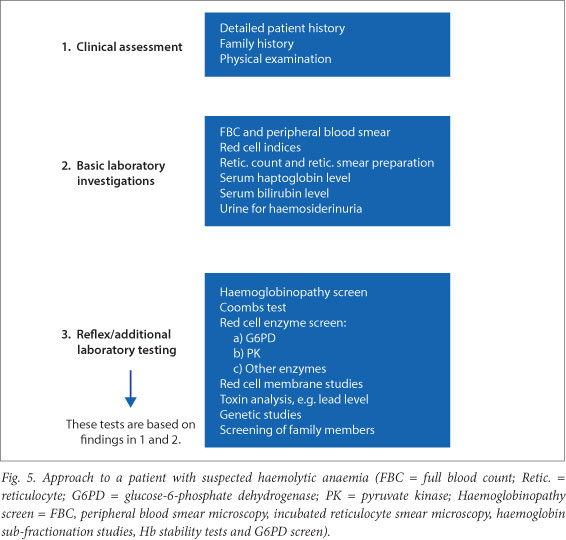
Fig. 5 outlines a generic approach to a patient with suspected haemolytic anaemia. The number of investigations is extensive and they should therefore be guided by the history and clinical examination. Initial basic investigations should be performed, followed by confirmatory and additional studies as indicated.
Conclusion
Haemoglobinopathies may present with sequelae of chronic haemolysis. A diagnostic approach in a patient with a suspected haemoglobinopathy requires a thorough history and clinical examination, directed laboratory testing including family studies, and an accurate interpretation of the results.
References
1. Bain BJ. Haemoglobinopathy Diagnosis. 2nd ed. London: Blackwell, 2001:2637,170-173. [ Links ]
2. Sarnaik S. Thalassemia and related hemoglobinopathies. Indian J Pediatr 2005;72(4):319-324. [ Links ]
3. Hoffbrand AV, Moss PAH. Essential Haematology. 6th ed. Wiley-Blackwell; John Wiley & Sons, 2011:89-97. [ Links ]
4. Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease: Life expectancy and risk factors for early death. N Engl J Med 1994;330(23):1639-1644. [http://dx.doi.org/10.1056/NEJM199406093302303] [ Links ]
5. Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease - rates and risk factors. N Engl J Med 1991;325(1):11-16. [http://dx.doi.org/10.1056/NEJM199107043250103] [ Links ]
6. Serjeant GR, Ashcroft MT, Serjeant BE, Milner PF. The clinical features of sickle-cell β thalassaemia in Jamaica. Br J Haematol 1973;24(1):19-30. [http://dx.doi.org/10.1111/j.1365-2141.1973.tb05723.x/pdf] [ Links ]
7. Yates A, Mortier N. The diagnostic dilemma of congenital unstable hemoglobinopathies. Pediatric Blood Cancer 2010;55(7):1393-1395. [http://dx.doi.org/10.1002/pbc.22702/pdf] [ Links ]
 Corresponding authors: R von Fintel (rosevonfintel@yahoo.co.uk), N A Alli (nazeer.alli@nhls.ac.za)
Corresponding authors: R von Fintel (rosevonfintel@yahoo.co.uk), N A Alli (nazeer.alli@nhls.ac.za)


{kind=link}