










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Child Health
versión On-line ISSN 1999-7671
versión impresa ISSN 1994-3032
S. Afr. j. child health vol.7 no.2 Pretoria may. 2013
CASE REPORT
Langerhans cell histiocytosis: A case presentation and literature review
K RautenbachI; D K StonesII
IMB ChB, MMed (Paed), Cert Med Oncol (Paed). Paediatric Haematology Oncology Unit, Universitas Hospital, Bloemfontein, South Africa
IIMB ChB, DCH (SA), FCPaed (SA), MMed (Paed). Paediatric Haematology Oncology Unit, Universitas Hospital, Bloemfontein, South Africa
ABSTRACT
Langerhans cell histiocytosis (LCH) is a disease of unknown aetiology. It is characterised by extreme clinical heterogeneity that can bring it to the attention of a variety of healthcare workers. A high index of suspicion is key to early diagnosis. Once the diagnosis has been confirmed, all cases should be referred to a paediatric oncologist for risk stratification and further management. We present a case of a toddler with LCH with skeletal involvement, who primarily presented to an orthopaedic surgeon. Relevant knowledge about this condition is reviewed.
Case report
An 18-month-old boy was referred to an orthopaedic surgeon with a 2-week history of a limping gait, and a left mid-shaft tibial bone tumour as seen on magnetic resonance imaging (MRI). Tumour resection and a bone transplant were done. Histological examination confirmed the diagnosis of Langerhans cell histiocytosis (LCH). The resection was deemed to be complete, and a follow-up skeletal isotope scan was scheduled for 3 months later.
At this follow-up visit the patient was clinically well, but the skeletal isotope scan revealed areas of abnormally increased uptake in the anterior aspects of the 7th and 8th ribs on the right side. A computed tomography (CT) scan confirmed the presence of a bone-destroying tumour affecting these ribs. A rib resection was performed and once again histological examination confirmed LCH. The boy was referred to a paediatric oncologist for further management.
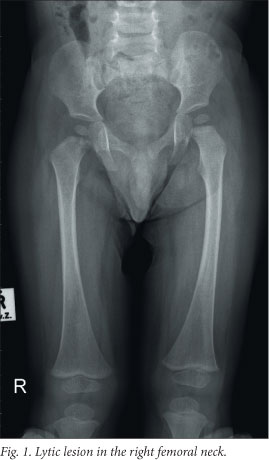
At the paediatric oncology unit a work-up was done to determine the extent of disease. Staging investigations included a complete blood count with differential counts, renal and liver function tests, measurement of ferritin and C-reactive protein, an endocrine profile, bone marrow examination, abdominal ultrasound, a chest radiograph and a skeletal survey. The staging investigations excluded any organ dysfunction. The skeletal survey showed lytic lesions in the left parasagittal area of the skull and the right femoral neck (refer to Fig. 1). A final diagnosis of LCH with multifocal skeletal involvement only was made.
Chemotherapy was commenced as per the protocol for multifocal bone disease. The initial intensive phase of chemotherapy consisted of daily prednisone and weekly vinblastine for a total of 6 weeks. Reevaluation imaging (skeletal survey, skeletal isotopes and CT of the facial bones) after the initial intensive phase showed that the patient still had active disease in some of the initially affected areas. A second initiation phase was therefore commenced. Imaging 6 weeks later confirmed that the patient now had non-active disease (i.e. regression of signs and symptoms and no new lesions). He therefore proceeded to the continuation phase of treatment, consisting of oral prednisone pulses and vinblastine every 3 weeks for a total of 52 weeks.
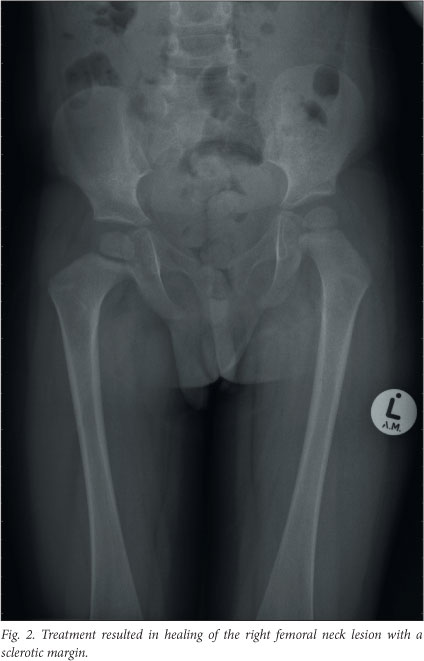
Imaging repeated just before the end of the continuation phase showed that all of the initial lesions had disappeared, but a new lesion was present on the medial aspect of the left ilium. The state of the disease was now defined as progressive or refractory. Since the new bony lesion was inaccessible, excluding curettage with intra-lesional steroids as an acceptable treatment option, second-line chemotherapy was commenced. The second-line treatment consisted of fortnightly methotrexate infusions, weekly vinblastine and daily oral prednisone, for a total of 6 weeks. At this stage the disease was in a regressive state and the patient proceeded to the second-line continuation chemotherapy consisting of 4 drugs (oral prednisone pulses, vinblastine every 3 weeks, weekly oral methotrexate and daily mercaptopurine). He successfully completed the 52 weeks of the continuation phase, and recent imaging confirmed that he still has non-active disease. The only remaining signs are a slight coxa vara deformity secondary to the right femoral neck scar (Fig. 2) and a localised depression in the lower ribcage at the site of the rib resection.
Discussion
LCH is a disease of unknown aetiology that is characterised by extreme clinical heterogeneity. Its clinical heterogeneity is illustrated by the fact that it was historically described as three different disease entities, based on disease extent and severity.[1-3] Letterer-Siwe disease is an acute disseminated and rapidly progressive form of LCH that is mostly seen in infants.[4] Besides potentially fatal visceral involvement, cutaneous lesions, often the presenting sign, are found in 80% of cases.[4] Between 1893 and 1920, Hand, Schüller and Christian elaborated the LCH triad of skull defects, exophthalmos and diabetes insipidus.[5] HandSchüller-Christian disease develops during early childhood and is a chronic, multi-focal form of LCH with an intermediate prognosis.[4] The least severe variant of LCH is mostly seen in older children and is called the eosinophilic granuloma. Lesions are either solitary or few, and mostly involve the bones.[4] Lesions are usually chronic with a tendency towards spontaneous healing. In 1953, the common pathology of these three conditions was recognised and the term histiocytosis X was introduced to name them collectively.[6] The letter X denoted the uncertainty about the pathogenesis of the disease.[6] About 2 decades later the cause was identified as being an abnormal proliferation of Langerhans cells, and the name Langerhans cell histiocytosis was adopted.[2] Today LCH is still the recommended term.[3,7]
Langerhans cells are histiocytes of dendritic cell lineage and constitute 2 - 4% of the normal resident epidermal cells.[4,5] Normal Langerhans cells express characteristic cell surface proteins called the CD1a protein and langerin. These cells can best be identified by their immunoreactivity for the antibodies to the CD1a antigen (anti-CD1a) and to langerin (anti-CD207).[3,4] Langerhans cells can be identified on electron microscopy by the presence of Birbeck granules.[3,4]
LCH is characterised histologically by an uncontrolled monoclonal proliferation of abnormal Langerhans cells: in contrast to their normal epidermal counterparts, LCH cells spread to almost any organ and accumulate there in a background of inflammatory cells, forming characteristic granulomas.[6] This clonal proliferation and infiltration may be localised or systemic, explaining the variable extent of involvement typical of LCH. Today most investigators consider LCH to result from an aberrant immune response, but the stimulus for clonal Langerhans cell proliferation remains unknown.[1,3] The definitive diagnosis of LCH is based on immunophenotypic and histological examination of lesional tissue.[2,5,8,9]
LCH is primarily a paediatric disease: it may affect any age group, but the peak incidence is at 1 - 4 years of age. It is a rare disease with an incidence of 3 - 5 per million children aged 1 - 14 years.[3-5,7,8] Cases are essentially sporadic.
The spectrum of presentation is broad and protean, ranging from a benign course to a fulminant progressive disease.[4,5,7] Almost any organ can be affected. According to the number of organs involved, it is possible to distinguish between single-system and multi-system disease.[3,7] The majority of patients have single-system disease, most cases involving skin or bone.[6] Multi-system disease consists of various combinations of multiple organ involvement, and there may also be organ dysfunction and pronounced generalised symptoms.[9]
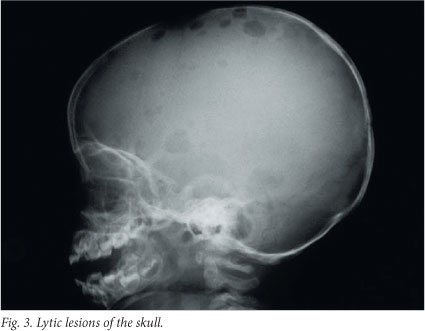
Bones are affected in 80% of cases - in fact, the clinical hallmark of LCH is lytic bone lesions. Any bone can be affected, but most commonly the skull (Fig. 3), pelvis, spine, mandible and ribs are involved.[2,6] The lesions may be occult, but usually present with pain or swelling. Skeletal X-rays are superior to skeletal isotopes in identifying the lesions. The radiographic appearance depends on the phase of the disease.[6] The early stage is characterised by an aggressive pattern of osteolysis and a periosteal reaction.[6,7] The late stage is characterised by a well-defined sclerotic margin.[6] Mandibular involvement gives the typical X-ray appearance of so-called floating teeth within the lytic lesion and is often associated with a soft-tissue swelling.[6] CT and MRI are the investigations of choice for evaluation of vertebral and craniofacial lesions.[7] Involvement of the orbital wall is a common manifestation and often results in proptosis.[6] Lesions of the facial bones or bones of the anterior or middle cranial fossae substantially increase the likelihood of underlying central nervous system involvement.[2,7]
Cutaneous involvement is encountered in 40% of cases.[4,7] It is often the first manifestation of LCH and is therefore of considerable diagnostic importance. It may be the only manifestation, especially in babies, but further investigation is mandatory to exclude more extensive disease.[6] LCH may cause different types of skin lesions, most often infiltrating nodules and plaques undergoing ulceration, but also scaly brown papules.[3,7] Infants may have a seborrhoeic scalp rash, which is often mistaken for true seborrhoea.[5,8] Seborrhoea-like involvement of the scalp may also resemble a severe case of dandruff. Skin involvement may be either localised or generalised and the scalp, abdomen and trunk are often affected.[4] When an infant or a young child presents with extensive, atypical or unremitting skin lesions (e.g. persistent diaper dermatitis) it should raise suspicion and LCH should be excluded histologically from a representative tissue sample.[4,10]
Multi-system LCH is characterised by many different possibilities of organ involvement, resulting in a great variety of clinical and radiological findings, none being pathognomonic in isolation. Histological diagnosis is mandatory. Haemopoietic involvement occurs most frequently in young children with diffuse disease and is defined as cytopenias, splenomegaly or bone marrow involvement.[7] Hepatic involvement is defined as hepatic enlargement, liver function test abnormalities and/or histological confirmation. Liver involvement may lead to hypo-albuminaemia with ascites, hyperbilirubinaemia and clotting factor deficiencies.[3] Lymph nodes are involved in less than 10% of patients. Gastro-intestinal involvement is found in less than 5%, and may lead to malabsorption syndromes.[6] A variety of CNS lesions have been described, ranging from hypothalamus-pituitary infiltration with secondary diabetes insipidus, which is another clinical hallmark of LCH, to eosinophilic granulomas and neurodegenerative disease (ataxia, cognitive dysfunction).[2,3,6-8]
Features of LCH that indicate a poor prognosis include age of less than 2 years at diagnosis, any risk organ involvement (bone marrow, blood, liver and spleen), organ dysfunction, multi-system disease and unsatisfactory response to initial treatment.[2,10] Children with poor prognostic features constitute less than 15% of patients with LCH. Patients without involvement of risk organs (low-risk group) are not at risk of death, but still need systemic therapy in order to control the disease activity and avoid reactivations and permanent consequences.
Irreversible long-term deficits that develop in areas of active LCH include small stature, growth hormone deficiency, hypothyroidism, hearing loss, cerebellar ataxia, orthopaedic deficits, biliary cirrhosis and portal hypertension.[3] Diabetes insipidus is the most frequent CNS-related permanent consequence in LCH, usually requiring lifelong replacement therapy. There is also an increased incidence of malignancy in long-term survivors.[4] Even though most LCH lesions eventually resolve spontaneously, the goal of therapy must be to rapidly achieve and maintain a non-active disease state in order to avoid the acute morbidity as well as the destructive sequelae that result from disease activity.
Since the pathogenesis of LCH is still unknown, the approach to treatment is empirical and not rational. With a malignant disease, the goal would be to eradicate the malignant clone. In contrast to malignant disease, the therapy of LCH aims to interrupt the cytokine expression that fuels the abnormal proliferation of LCH cells.[1,6] In view of its clinical heterogeneity, patients are risk-stratified at the time of diagnosis, based upon the extent of disease and whether or not risk organs are involved.[5,9] Because of its rarity, there are limited data with regard to the optimal treatment for patients with multisystem LCH, and participation in a well-designed clinical trial is strongly recommended.[1,3] There is general agreement that intensive systemic therapy (for a total of12 months) is indicated for all patients with multi-system LCH.[1,8] A concept exists that osseous LCH, even if multifocal, is a benign, self-limiting disease.[2] However, this may not necessarily apply to patients with other poor prognostic features, such as an age of less than 2 years. In patients with multiple bone lesions or multisystem non-risk organ involvement, even a short treatment with only a single agent (e.g. prednisone) is insufficient, since reactivations commonly occur in this group of patients. According to the Histiocyte Society LCH-III study, they should receive treatment with vinblastine and prednisone for at least 6 - 12 months.[3,9]
Conclusion
Because of its extreme clinical heterogeneity, LCH can present to a variety of specialists (e.g. general paediatricians, dentists, dermatologists and orthopaedic surgeons), and a high index of suspicion is necessary to make a timely diagnosis. The clinical hallmark of LCH is lytic bone lesions. LCH should also be considered in the case of idiopathic diabetes insipidus, or atypical or extensive skin lesions in infants or young children. An accurate histological diagnosis is needed. Any child with a suspected solitary LCH lesion must have a full diagnostic work-up to exclude multiple lesions. All patients with LCH must be referred to a paediatric oncologist for proper risk stratification, an appropriate treatment plan and long-term follow-up. Systemic therapy is indicated to prevent irreversible damage to normal tissue and long-term consequences.
References
1. Minkov M. Multisystem Langerhans cell histiocytosis in children: Current treatment and future directions. Pediatric Drugs 2011;13(2):75-86. [http://dx.doi.org/10.2165/11538540-000000000-00000] [ Links ]
2. Tokuç G, Boran P, Öktem S. Langerhans cell histiocytosis in Turkish children. Turk J Pediatr 2009;51(4):344-349. [ Links ]
3. McClain KL, Allen CE, Hicks J. Histiocytic diseases. In: Pizzo AP, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2011:703-716. [ Links ]
4. Hussein MRA. Skin-limited Langerhans cell histiocytosis in children. Cancer Invest 2009;27(5):504-511. [http://dx.doi.org/10.1080/07357900802216452] [ Links ]
5. Filipovich A, McClain K, Grom A. Histiocytic disorders: Recent insights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant 2010;16(1):S82-S89. [http://dx.doi.org/10.1016/j.bbmt.2009.11.014] [ Links ]
6. Kilborn TN, The J, Goodman TR. Paediatric manifestations of Langerhans cell histiocytosis: A review of the clinical and radiological findings. Clin Radiol 2003;58(4):269-278. [http://dx.doi.org/10.1016/S0009-9260(02)00537-8] [ Links ]
7. McClain KL. Clinical manifestations, pathological features and diagnosis of Langerhans cell histiocytosis. http://www.uptodate.com (accessed August 2012). [ Links ]
8. Schmidt S, Eich G, Hanquinet S, et al. Extra-osseous involvement of Langerhans cell histiocytosis in children. Pediatr Radiol 2004;34(4):313-321. [http://dx.doi.org/10.1007/s00247-003-1118-z] [ Links ]
9. McClain KL. Treatment of Langerhans cell histiocytosis. http://www.uptodate.com (accessed August 2012). [ Links ]
10. Lau L, Krafchik B, Trebo MM, et al. Cutaneous Langerhans cell histiocytosis in children under one year. Pediatr Blood Cancer 2006;46(1):66-71. [http://dx.doi.org/10.1002/pbc.20479] [ Links ]
 Corresponding author: K Rautenbach (RautenbachK.MD@ufs.ac.za)
Corresponding author: K Rautenbach (RautenbachK.MD@ufs.ac.za)

