










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkWater SA
On-line version ISSN 1816-7950
Print version ISSN 0378-4738
Water SA vol.42 n.3 Pretoria Jul. 2016
http://dx.doi.org/10.4314/wsa.v42i3.09
ORIGINAL ARTICLES
Catalytic wet peroxide oxidation of formic acid in wastewater with naturally-occurring iron ore
E ErasmusI, *; JO ClaassenII; WA van der WesthuizenII
IDepartment of Chemistry, University of the Free State, Bloemfontein, 9300, Republic of South Africa
IIDepartment of Geology, University of the Free State, Bloemfontein, 9300, Republic of South Africa
ABSTRACT
The catalytic wet oxidation of formic acid, using hydrogen peroxide as the oxidizing agent over naturally-occurring iron ore, was explored. Firstly, the decomposition of hydrogen peroxide to its hydroxyl radicals (HO- and HOO-) over naturally-occurring iron ore was investigated. The reaction was monitored by ATR FTIR by following the disappearance of the O-H peak of H2O2 at 2 860 cm-1. Decomposition occurred according to the Fenton mechanism and resulted in observed first-order rate constants one order of magnitude faster than that without the catalyst. Turnover frequencies (TOF) of 1.97-8.85 x 10-9 s-1 were obtained for the decomposition of H2O2. The wet oxidation of formic acid using hydrogen peroxide as the oxidizing agent over naturally-occurring iron ore reaction was also monitored by ATR FTIR, following the disappearance of the carbonyl stretching frequency of formic acid at 1 727 cm-1. Experiments were performed at different hydrogen peroxide (2, 4, 6 and 8M) and formic acid (1.26, 2.52, 6.3 and 12.6 M) concentrations as well as with varying amounts of naturally-occurring iron ore catalyst, at pH = 2. Elevated hydrogen peroxide and formic acid concentrations led to increased observed first-order kinetics, as high as kobs= 21.75 x 104 min1 with a TOF = 1.73 x 10-8 - 1.12 x 10-6 s-1.
Keywords: iron ore, catalytic wet oxidation, formic acid, Fenton, hydrogen peroxide
INTRODUCTION
Apart from the large-scale exploitation of iron containing ore as raw material feed for iron extraction, it is one of the few naturally-occurring minerals that have been exploited for alternative applications. As a result, numerous studies report on the use of iron-containing minerals as adsorbents (Nowack et al., 2006; Rovira et al., 2008; Zeng et al., 2004; Spiteri et al., 2008; Guo et al., 2007; Zhang et al., 2004; Pirillo et al., 2009) and catalysts (Costa et al., 2008; Huang et al., 2000; Centi et al., 2000; Andreozzi et al., 2003; Du et al., 2008) for the removal of pollutants from wastewaters.
Formic acid is a pollutant in wastewater from the effluents of some industries (Claudel et al., 1984). It is also a stable intermediate from the decomposition of many organic pollutants (Chou et al., 1998; Davis et al., 1999; Hwang et al., 1999; Dinsdale et al., 2000), which is difficult to oxidize further (Ogata et al., 1981; Harmsen et al., 1997). For this reason, formic acid is often used as a model organic pollutant in wastewater treatment studies (Kawaguchi et al., 1993; Kim et al., 1996; Butterfield et al., 1997; Candal et al., 1997; Miller et al., 1999).
Wet air oxidation is a procedure commonly used to reduce the total organic carbon (TOC) in industrial wastewater (Matatov-Meytal et al., 1998; Mishra et al., 1995; Levec et al., 1995; Luck et al., 1996; Gallezot et al., 1997; Klinghoffer et al., 1998; Romalho et al., 1983.). The reaction however requires high operating temperatures. The use of oxidation agents allows the process to precede at lower reaction temperatures. Ozone and hydrogen peroxide (H2O2) are the preferred oxidants. These compounds are active even at room temperature and decompose to environmentally friendly O2 and H2O.
Water treatment with ozone is well known (Masten et al., 1994; Luck et al., 1997; Allemane et al., 1993; Andreozzi et al., 1996). Its effectiveness is however limited by lower solubility at atmospheric pressure and the short lifetime of ozone renders the process more expensive. H2O2 is therefore the preferred oxidant.
The use of hydrogen peroxide to oxidize the pollutants in wastewater requires it to be in its radical form. The production of hydroxyl radicals from H2O2 and minimization of its decomposition to water is achieved either photochemically or by catalysis. The photochemical activation of hydrogen peroxide requires UV-radiation. However, due to its low extinction coefficient, the operation process is costly (Ojima et al., 1994). The formation of the hydroxyl radicals using iron ion catalysis is well known (Edwards et al., 1992; Walling et al., 1975). The process proceeds according to the Fenton mechanism as illustrated below (Centi et al., 2000):
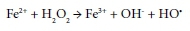
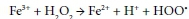
Numerous examples are available for the oxidation of different organic pollutants in wastewater, using a combination of iron ion and hydrogen peroxide. There are however very few publications available on the use of naturally-occurring iron ore (such as hematite, ferrihydrite, goethite and lepidocrocite) as a cheap alternative for the oxidation of pollutants such as formic acid in wastewater.
This study utilizes hard high-grade hematite ore as such an alternative. The ore is massive Gamagaran type ore from the Sishen Iron Formation in the Northern Cape Province of the Republic of South Africa. It comprises sand to silt size hematite grains in a matrix which appears as ferruginized, hematitic sandstone and shale. Small amounts of specular hematite are also present (Wilson et al., 1998).
EXPERIMENTAL
Chemicals and instruments
The hematite ore used in this study was sourced from Sishen Iron Ore Mine (see Fig. 1). The hematite was crushed and screened with only the particles in the size range 75-212 μm used. All reagents (Merck, Sigma-Aldrich) were used without further purification and water was double distilled. Attenuated total reflectance Fourier transform infrared (ATR FTIR) spectra were recorded on a Nicolet IS 50 infrared spectrophotometer equipped with a diamond ATR. X-ray photoelectron spectroscopy (XPS) data were recorded on a PHI 5000 Versaprobe system, with monochromatic Al Kcx X-ray source. Spectra were obtained, using the aluminium anode (Al Kcx = 1486.6 eV), operating at 50 μm, 12.5 W and 15 kV energy. The survey scans were recorded at constant pass energy of 187.85 eV. The detailed regional scans were recorded at constant pass energy of 29.35 eV and analyser resolution of < 0.5 eV. The background pressure was 2 x 10-8 Pa. The XPS data were analysed with Multipak version 8.2c computer software (Moulder et al., 1995), applying Gaussian-Lorentz fits (the Gaussian/Lorentz ratios were always > 95%). The iron ore were characterized by X-ray diffraction (XRD) using a Panalytical Empyrean diffractometer for elemental analysis.

Hydrogen peroxide decomposition test without the catalyst
2M hydrogen peroxide solutions in water were prepared according to Table 2, while stirring at room temperature. The pH was adjusted with the addition of either H2SO4 or NaOH similar to published procedures (Li, 1999). The final test volume was 10 mL. Samples were taken at regular intervals and analysed by ATR FTIR.
Hydrogen peroxide decomposition test with the naturally-occurring iron ore added as catalyst
Hydrogen peroxide reaction mixtures of different concentrations (2, 4, 6 and 8 M) were prepared in water according to Table 2, while stirring at room temperature. After the pH was adjusted with the addition of H2SO4 to obtain a pH of 2, 20 mg crushed Fe2O3 ore was added to the reaction mixture. Samples were taken at regular intervals and analysed by ATR FTIR.
Hydrogen peroxide decomposition and formic acid oxidation test with Fe2O3 catalyst

Reaction mixtures (10 mL) were prepared according to Table 3, while stirring at 35°C. After the pH was adjusted to 2 with the addition H2SO4, the required amounts of formic acid and Fe2O3 catalyst (iron ore) were added. Samples were taken at regular intervals and analysed by ATR FTIR.
Turnover frequencies (TOF) were determined by the following equation:
In addition, pseudo first-order rate constants, koobs, were calculated by fitting kinetic data to the first-order equation
(Espenson, 1995) [A]t = [A]0 e(kobsi) with [A]t and [A]0 the absorbance of the indicated species at time t and at t = 0.
Preliminary stability testing of the natural iron ore
A 50 mg sample of the naturally-occurring iron ore was stirred in a solution of 4 M hydrogen peroxide at pH 2 for 10 days. This test showed that no iron leaching occurred during the time period, which was the same as was used for the catalytic testing.
RESULTS AND DISCUSSION
Chemical analysis of the iron ore
XRD analysis of the iron ore revealed that the sample contains 98.13% Fe2O3, 0.45% Al2O3 and 1.12% SiO2.
X-ray photoelectron spectroscopy
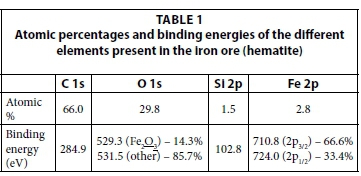
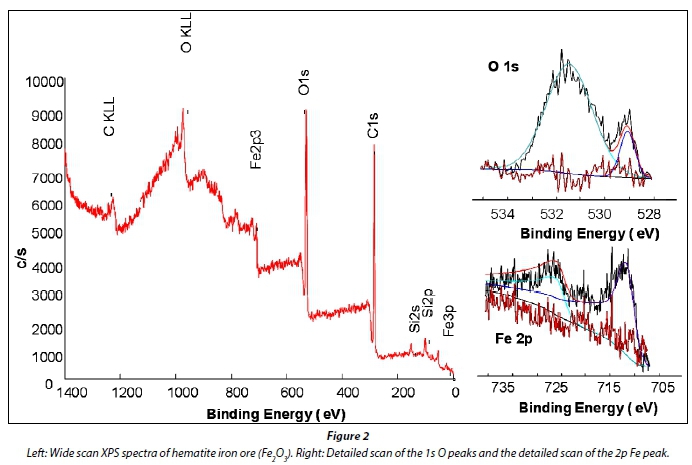
The XPS spectra of the iron ore (Fig. 2) revealed that the sample contained Fe, O, C (possibly from CaCO3) and Si. Table 1 summarizes the atomic percentages and binding energies (eV) of these elements. XPS samples most often exhibit adventitious carbon contamination which is detected by XPS analysis (Castaneda et al., 2010; Kyotani et al., 1997). The binding energy of these carbonaceous deposits is set at 284.9 eV and the other element's binding energies are referenced against it. The detected silicon is most probably from mineral contaminants such as quartz, as indicated by the XRD analysis.
The binding energy of the Fe 2p3/2 (see Fig. 2 right) was detected at 710.8 eV, which is in agreement with published values of 710.8 eV for Fe2O3 (Brion, 1980; Nefedov et al., 1975). The detailed scan of the oxygen 1s area (see Fig. 2 right) showed that there are two distinct peaks present. The peak observed at 531.5 eV, which accounts for 85.7% of the oxygen present in the sample, is from adsorbed oxygen and other adventitious sources, normally present in all samples. The peak observed at 529.3 eV, which accounts for 14.3% of the oxygen present, is allocated to oxygen present in iron oxide Fe2O3. When calculated, the peak at 529.3 eV accounts for 4.3% of the total atomic percentage. This value is in agreement with the amount of oxygen expected if the iron content in Fe2O3 is 2.8%. The binding energy of the O 1s (529.3 eV) in Fe2O3, is in close agreement with published values of 529.5 and 529.6 eV for Fe2O3 (Haber et al., 1976; Nefedov et al., 1977).
Decomposition of hydrogen peroxide
Hydrogen peroxide is a thermodynamically unstable compound that decomposes to form oxygen and water. Various factors influence the rate of decomposition, including temperature and pH. The decomposition of hydrogen peroxide is enhanced when catalysts such as Pt and MnO2 are added (Petrucci, 2007). However when certain metal ions, including Fe2+, are used as a catalyst the decomposition proceeds according to the Fenton mechanism to produce the free radicals HO' and HOO'.
Since this study is concerned with the decomposition of hydrogen peroxide using naturally-occurring iron ore, it is also important to investigate the decomposition of hydrogen peroxide in the absence of iron ore.
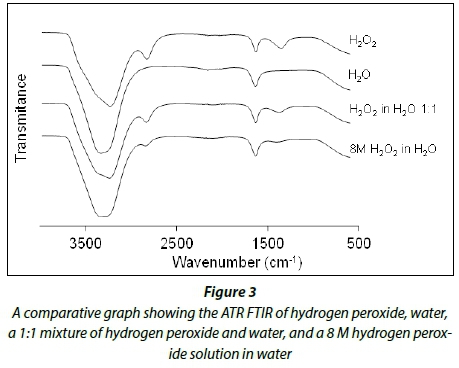
Figure 3 shows the comparative ATR FTIR graphs of hydrogen peroxide, water, a 1:1 mixture of hydrogen peroxide and water, and an 8 M hydrogen peroxide solution in water. The distinctive frequencies associated with water are the -OH bending frequency at 1 646 cm-1 and the -OH stretching frequency in the area 2 800-3 800 cm-1. Hydrogen peroxide shows 4 peaks: 1 370, 1 646, 2 860 cm-1, and 2 800-3 800 cm-1. These peaks are assigned to the asymmetric deformation band of H2O2 (v1), the -OH bending (v2), a combination of v1, v2 and v3, and the -OH stretching frequency (v3), respectively. These assignments are in agreement with published data from Giguere (1950). The peak observed at 2 860 cm-1 was used to follow the decomposition of hydrogen peroxide.
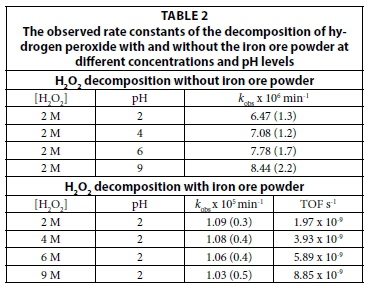
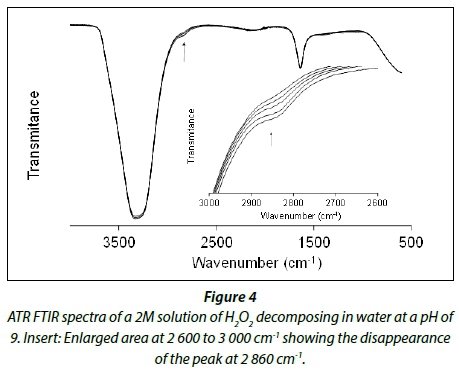
The decomposition of 2M hydrogen peroxide solutions in water was followed by ATR FTIR at different pH levels. Figure 4 shows the ATR FTIR spectra of the decomposition of a 2 M solution of hydrogen peroxide in water at a pH of 9. It is known from literature that hydrogen peroxide is more stable at lower pH levels, as indicated in Table 2. Thus the rest of the experiments were conducted at a pH of 2 to keep self-decomposition to a minimum.
The decomposition of hydrogen peroxide solutions of different concentrations (2, 4, 6 and 8 M) were followed by ATR FTIR at pH = 2 in the presence of naturally-occurring iron ore (Fe2O3).
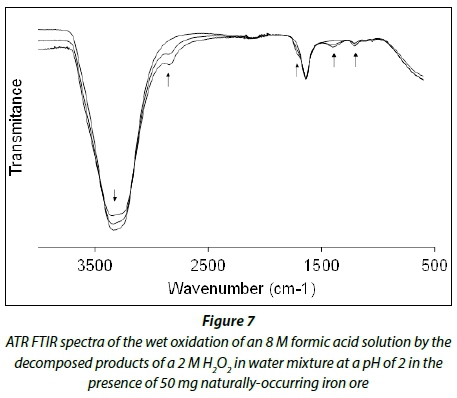
Figure 5 depicts the ATR FTIR spectra of the decomposition of an 8 M solution of hydrogen peroxide in water at a pH of 2 in the presence of a catalytic amount (20 mg) of naturally-occurring iron ore.
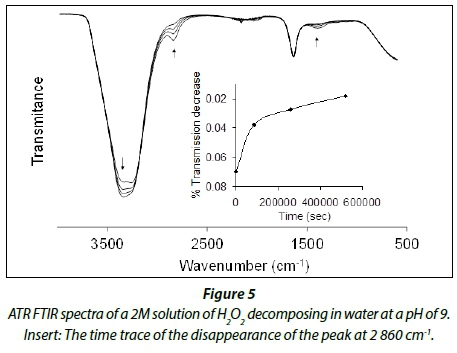
The observed rate constants obtained for the different concentrations of hydrogen peroxide (see Table 2) are, within experimental conditions, the same, at approx. 1.0 x 10-5min-1. This is approximately one order of magnitude larger than values observed for the decomposition of hydrogen peroxide in the absence of iron ore. The slower than expected rate of decomposition of the hydrogen peroxide in the presence of the iron ore can be attributed to, firstly, the small amount of active catalyst (the small amount of Fe2O3 at the surface of the powdered sample available for catalysis). Secondly, it is known that iron in its oxide form is a more ineffective catalyst for the decomposition of hydrogen peroxide than other iron ions such as FeCl2, FeCl3, Fe(NO3)3, etc. Lastly, the low solubility of the iron ore at the chosen conditions affects diffusion during the reaction.
Iron oxide may be a relatively slow catalyst for the decomposition of hydrogen peroxide. Since the aim of the study is to catalytically oxidize formic acid using the hydroxyl radical, fast decomposition of the hydrogen peroxide is not required.
The observed rate of hydrogen peroxide decomposition over naturally-occurring iron ore (measured in this study) compares well with studies conducted on hematite (0.9 x 10-5 min-1) and other iron-containing catalysts reported in literature (Huang et al., 2001).
Catalytic wet oxidation of formic acid
Figure 6 depicts the comparative ATR FTIR graphs of formic acid and a variety of different concentrations (2, 4, 6 and 8 M) of formic acid dissolved in water at pH = 2. The arrows (in the bottom spectra) indicate how the peaks increase or decrease as the concentration of the formic acid is increased. The distinctive frequencies observed for formic acid dissolved in water appear at 2 856, 1 727, 1 413 and 1 200 cm-1, representing the C-H stretching frequency (v C-H), the C=O stretching frequency (v C=O) and the two CH2 bending frequency (u>CH2), respectively. These assignments were made in agreement with published data from Popova et al. (2000).
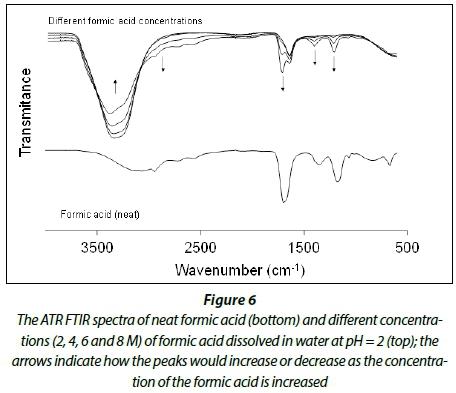
Since the C-H stretching frequency of formic acid overlaps with the distinctive combination peaks (v1, v2 and v3) of H2O2 at 2 860 cm-1, the C=O stretching frequency at 1 727 cm-1 was used to follow the oxidation of formic acid in the following experiments.
The wet oxidation of formic acid by decomposed hydrogen peroxide radicals (HO' and HOO') over naturally-occurring iron ore solutions was followed by ATR FTIR using different concentrations of hydrogen peroxide (2, 4, 6 and 8 M) and formic acid (1.26, 2.52, 6.3 and 12.6 M) as well as varying amounts of Fe2O3 catalyst at pH = 2. Figure 7 shows the ATR FTIR spectra of the wet oxidation of an 8 M formic acid solution by the hydroxyl radicals (HO' and HOO') from decomposed 2 M H2O2 in water at a pH of 2 in the presence of a catalytic amount of naturally-occurring iron ore. The different experimental conditions and its observed first-order rate constants are presented in Table 3. These observed rate constants are two orders of magnitude larger than that observed for the decomposition of hydrogen peroxide. This could be attributed to an increased amount of catalyst used and the formation of two hydroxyl radicals that can react with the formic acid.
From Table 3 (Experiments A1-A6) and Fig. 8, it is evident that the decomposition rate increases with an increase in the hydrogen peroxide concentration. This is expected since higher concentrations of hydrogen peroxide will result in the formation of more hydroxyl radicals, which in turn would oxidize the formic acid faster.
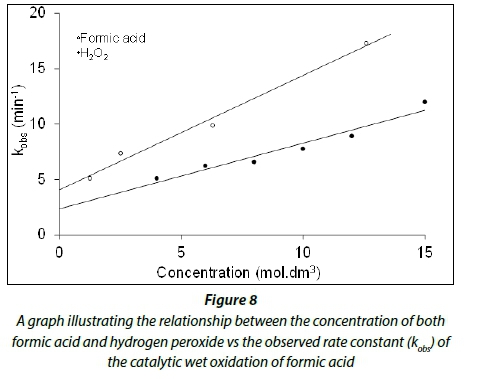
Experiments A1, B1, C1 and D1, where the hydrogen peroxide concentration and amount of iron ore catalyst are kept constant while the concentration of the formic acid is being varied, indicated that the rate of decomposition also increases as the concentration of the formic acid increases (see Fig. 8). More formic acid molecules present in solution would lead to more interaction between the formic acid and the hydroxyl molecules which would in turn lead to a faster formic acid oxidation rate being observed.
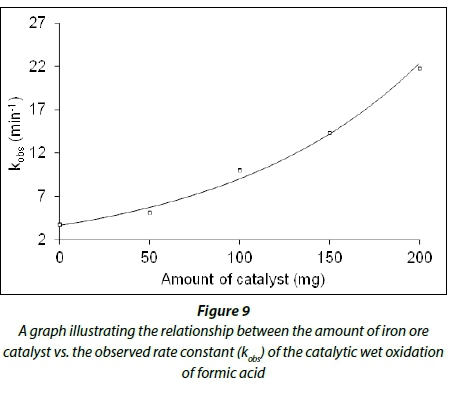
In the next set of experiments, A1 and A7-9, the hydrogen peroxide and formic acid concentrations were kept constant while the amount of catalyst was varied (see Table 3). From Fig. 9, it can be seen that the observed rate constant for the catalytic wet oxidation of formic acid exponentially increases as the amount of catalyst increases.
CONCLUSION
It has been confirmed that formic acid can be effectively wet oxidized by the decomposed products of hydrogen peroxide over a naturally-occurring iron ore catalyst. The decomposition of hydrogen peroxide to its hydroxyl radicals (HO' and HOO') is influenced by the pH of the solution. At higher pH, hydrogen peroxide is more unstable and decomposition can occur spontaneously. At a constant pH of 2, an increase in the concentration of hydrogen peroxide in aqueous solutions does not influence the rate of catalytic decomposition over naturally-occurring iron ore with an approx. kobs= 1 x 105 min1 observed under the chosen experimental conditions.
The catalytic wet oxidation of formic acid by decomposed hydrogen peroxide radicals (HO' and HOO') over naturally-occurring iron ore indicated that higher levels of hydrogen peroxide and formic acid concentration result in higher formic acid oxidation rates. This can be attributed to increased amounts of reacting molecules present in solution.
ACKNOWLEDGEMENTS
The authors acknowledge the financial support provided by the University of the Free State.
REFERENCES
ALLEMANE H, DELOUANE B, PAILLARD H and LEGUBE B (1993) Drinking water treatment with ozone. Ozone Sci. Eng. 15 419-424. http://dx.doi.org/10.1080/01919512.1993.10555733 [ Links ]
ANDREOZZI R, CAPRIO V and MAROTTA R (2003) Iron(III) (hydr) oxide-mediated photooxidation of 2-aminophenol in aqueous solution: a kinetic study. Water Res. 37 3682-3688. http://dx.doi.org/10.1016/S0043-1354(03)00271-9 [ Links ]
ANDREOZZI R, INSOLA A, CAPRIO V, MAROTTA R and TUFANO V (1996) The use of manganese dioxide as a heterogeneous catalyst for oxalic ozonation in aqueous solution Appl. Catal. A: Gen. 138 75-81. http://dx.doi.org/10.1016/0926-860X(95)00247-2 [ Links ]
BRION D (1980) Etude par spectroscopie de photoelectrons de la degradation superficielle de FeS2, CuFeS2, ZnS et PbS a lair et dans l'eau. Appl. Surf. Sci. 5 133-152. http://dx.doi.org/10.1016/0378-5963(80)90148-8 [ Links ]
BUTTERFIELD IM, CHRISTENSEN PA, HAMNETT A, SHAW KE, WALKER GM, WALKER SA and HOWARTH CR (1997) Applied studies on immobilised titanium dioxide films as catalysts for pho- toelectrochemical detoxification of water. J. Appl. Electrochem. 27 385-395. http://dx.doi.org/10.1023/A:1018453402332 [ Links ]
CANDAL RJ, ZELTNER WA and ANDERSON MA (1997) Titanium-supported titania photoelectrodes made by sol-gel processes. J. Environ. Eng. 125 906-912. http://dx.doi.org/10.1061/(ASCE)0733-9372(1999)125:10(906) [ Links ]
CASTANEDA CM, ASHBAUGH L and WAKABAYASHI P (2010) Use of proton backscattering to determine the carbon and oxygen content in fine particle samples deposited on PTFE((CF2)(n)) membrane disk filters. J. Aerosol Sci. 41 99-107. http://dx.doi.org/10.1016/j.jaerosci.2009.08.011 [ Links ]
CENTI G, PERATHONER S, TORRE T and GRAZIA VERDUNA M (2000) Catalytic wet oxidation with H2O2 of carboxylic acids on homogeneous and heterogeneous Fenton-type catalysts. Catal. Today 55 61-69. http://dx.doi.org/10.1016/S0920-5861(99)00226-6 [ Links ]
CLAUDEL B, NUEILATI M and ANDRIEU J (1984) Oxidation of formic-acid in aqueous-solution by palladium catalysts. Appl. Catal. 11 217-225. http://dx.doi.org/10.1016/S0166-9834(00)81880-1 [ Links ]
CHOU S, HUANG Y-H, LEE S-N, HUANG G-H and HUANG C (1998) Treatment of high strength hexamine-containing wastewater by electro-Fenton method. Water Res. 33 751-759. http://dx.doi.org/10.1016/S0043-1354(98)00276-0 [ Links ]
COSTA RCC, MOURA FCC, ARDISSON JD, FABRIS JD and LAGO RM (2008) Highly active heterogeneous Fenton-like systems based on Fe0/Fe3O4 composites prepared by controlled reduction of iron oxides. Appl. Catal., B: Environ. 83 131-139. http://dx.doi.org/10.1016/j.apcatb.2008.01.039 [ Links ]
DAVIS AP and GREEN DL (1999) Photocatalytic oxidation of cadmium-EDTA with titanium dioxide. Environ. Sci. Technol. 33 609-617. http://dx.doi.org/10.1021/es9710619 [ Links ]
DINSDALE RM, HAWKES FR and HAWKES DL (2000). Biodegradation of short organic acids in an expanded granular sludge bed reactor. Water Res. 34 2433-2438. http://dx.doi.org/10.1016/S0043-1354(99)00430-3 [ Links ]
DU W, XU Y and WANG Y (2008) Photoinduced degradation of orange II on different iron (hydr)oxides in aqueous suspension: Rate enhancement on addition of hydrogen peroxide, silver nitrate, and sodium fluoride. Langmuir 24 175-181. http://dx.doi.org/10.1021/la7021165 [ Links ]
EDWARDS JO and CURCI R (1992) Catalytic Oxidations with Hydrogen Peroxide as Oxidant. Kluwer, Dordrecht. [ Links ]
ESPENSON JH (1995) Chemical Kinetics and Reaction Mechanisms (2nd edn). McGraw-Hill, New York. [ Links ]
GALLEZOT P, CHAUMET S, PERRARD A and ISNARD P (1997) Catalytic wet air oxidation of acetic acid on carbon-supported ruthenium catalysts J. Catal. 168 104-109. http://dx.doi.org/10.1006/jcat.1997.1633 [ Links ]
GIGUERE PA (1950) The infra-red spectrum of hydrogen peroxide J. Chem. Phys. 18 88-92. http://dx.doi.org/10.1063/L1747464 [ Links ]
GUO H, STÜBEN D and BERNER Z (2007) Removal of arsenic from aqueous solution by natural siderite and hematite. Appl. Geochem. 22 1039-1051. http://dx.doi.org/10.1016/j.apgeochem.2007.01.004 [ Links ]
HABER J, STOCH J and UNGIER L (1976) X-ray photoelectron spectra of oxygen in oxides of Co, Ni, Fe and Zn J. Electron Spectrosc. Relat. Phenom. 9 459. http://dx.doi.org/10.1016/0368-2048(76)80064-3 [ Links ]
HARMSEN JMA, JELEMENSKY L, VAN PJM, KUSTER BFM and MARIN GB (1997) Kinetic modeling for wet air oxidation of formic acid on a carbon supported platinum catalyst Appl.Catal. A. 165 499-509. http://dx.doi.org/10.1016/S0926-860X(97)00232-9 [ Links ]
HUANG HH, LU MC and CHEN JN (2001) Catalytic decomposition of hydrogen peroxide and 2-chlorophenol with iron oxides. Water Res. 35 2291-2299. http://dx.doi.org/10.1016/S0043-1354(00)00496-6 [ Links ]
HWANG D-S, LEE E-H, KIM K-W, LEE K-I and PARK S-J (1999) Denitration of simulated high-level liquid waste by formic acid. J. Ind. Eng. Chem. 5 45-51. [ Links ]
KAWAGUCHI H (1993) Photo-oxidation of formic acid in aqueous solution in the presence of hydrogen peroxide. Chemosphere 26 1965-1970. http://dx.doi.org/10.1016/0045-6535(93)90023-X [ Links ]
KIM DH, ANDERSON MA (1996) Solution factors affecting the photocatalytic and photoelectrocatalytic degradation of formic acid using supported TiO2 thin films J. Photochem. Photobiol. 94 221-229. http://dx.doi.org/10.1016/1010-6030(95)04178-8 [ Links ]
KLINGHOFFER AA, CERRO RL, ABRAHAM MA (1998) Catalytic wet oxidation of acetic acid and using platinum on alumina monolith catalyst Catal. Today 40 59-71. http://dx.doi.org/10.1016/S0920-5861(97)00122-3 [ Links ]
KYOTANI T, NAGAI T, INOUE S, TOMITA (1997) Formation of new type of porous carbon by carbonization in zeolite nanochannels Chem. Mater. 9 609-615. http://dx.doi.org/10.1021/cm960430h [ Links ]
LEVEC J, PINTAR A (1995) Catalytic-Oxidation of Aqueous Solutions of Organics - An Effective Method for Removal of Toxic Pollutants From Watse-Water Catal. Today 24 51-58. http://dx.doi.org/10.1016/0920-5861(95)00006-2 [ Links ]
LI Y-S (1999) The use of waste basic oxygen furnace slag and hydrogen peroxide to degrade 4-chlorophenol. Waste Management 19 495-502. http://dx.doi.org/10.1016/S0956-053X(99)00239-1 [ Links ]
LUCK F, DJAFER M, LEITNER NKV, GOMBERT B and LEGUBE B (1997) Destruction of pollutants in industrial rinse waters by advanced oxidation processes. Water Sci. Technol. 35 287-292. http://dx.doi.org/10.1016/S0273-1223(97)00037-1 [ Links ]
LUCK F (1996) A review of industrial catalytic wet air oxidation processes. Catal. Today 27 195-202. http://dx.doi.org/10.1016/0920-5861(95)00187-5 [ Links ]
MASTEN SJ and DAVIES SHR (1994) The use of ozonization to degrade organic contaminants in wastewaters. Environ. Sci. Technol. 28 181-185. http://dx.doi.org/10.1021/es00053a001 [ Links ]
MATATOV-MEYRAL YI and SCHEINTUCH M (1998) Catalytic abatement of water pollutants. Ind. Eng. Chem. Res. 37 309-326. http://dx.doi.org/10.1021/ie9702439 [ Links ]
MISHRA VS, MAHAJANI VV and JOSHI JB (1995) Wet air oxidation. Ind. Eng. Chem. Res. 34 2-48. http://dx.doi.org/10.1021/ie00040a001 [ Links ]
MILLER LW, TEJEDOR-TEJEDOR MI and ANDERSON MA (1999) Titanium dioxide-coated silica waveguides for the photocatalytic oxidation of formic acid in water. Environ. Sci. Technol. 33 2070-2075. http://dx.doi.org/10.1021/es980817g [ Links ]
MOULDER F, STICKLE WF, SONOL PE and BOMBEN KD (1995) Handbook of X-ray Photoelectron Spectroscopy. ULVAC-PHI, Inc. Japan, Chigasaki. [ Links ]
NEFEDOV VI, GATI D, DZHURINSKII BF, SERGUSHIN NP and SALYN YV (1975) Simple and coordination compounds. Zh. Neorf. Khimii. 20 2307-2314. [ Links ]
NOWACK B, STONE AT (2006) Competitative adsorption of phosphate and phosphonates onto goehite. Water Res. 40 2201-2209. http://dx.doi.org/10.1016/j.watres.2006.03.018 [ Links ]
OJIMA S, ABE M and YAMAKI Y (1994) A new system for high-purity water production. Ultrapure Water 11 45-48. [ Links ]
OGATA Y, TOMIZAWA K, TAKAGI K (1981) Photooxidation of formic, acetic, and propionic acids with aqueous hydrogen peroxide. Can. J. Chem. 59 14-18. http://dx.doi.org/10.1139/v81-003 [ Links ]
PIRILLO S, FERREIRA ML, RUEDA EH (2009) The effect of pH in the adsorption of alizarin and eriochrome blue black R onto iron oxides. J. Hazardous Mater. 168 168-178. http://dx.doi.org/10.1016/j.jhazmat.2009.02.007 [ Links ]
ROVIRA M, GIMENEZ J, MARTINEZ M, MARTINEZ-LLADO X, PABLO J, MARTI V, DURO L (2008) Sorption of selenium(IV) and selenium(VI) onto natural iron oxides: Geothite and hematite. J. Hazardous Mater. 150 279-274. http://dx.doi.org/10.1016/j.jhazmat.2007.04.098 [ Links ]
ROMALHO RS (1983) Introduction to Wastewater Treatment Processes (2nd edn). Academic Press Publisher, New York. [ Links ]
SPITERI C, CAPPELLEN PV and REGNIER P (2008) Surface complexation effects on phosphate adsorption to ferric iron oxyhydroxides along pH and salinity gradients in estuaries and coastal aquifers. Geohim. Cosmochim. Acta 72 3431-3445. http://dx.doi.org/10.1016Aj.gca.2008.05.003 [ Links ]
WALLIN GC (1975) Fenton's reagent revisited. Acc. Chem. Res. 8 125-131. http://dx.doi.org/10.1021/ar50088a003 [ Links ]
WILSON MGC and ANHAEUSSER CR (1998) The Mineral Resources of South Africa, Handbook 16 (6th edn). Council for Geoscience, Pretoria. [ Links ]
ZENG L, LI X and LIU J (2004) Adsorptive removal of phosphate from aqueous solutions using iron oxide tailings. Water Res. 38 1318-1326. http://dx.doi.org/10.1016/j.watres.2003.12.009 [ Links ]
ZHANG W, SINGH P, PALING E and DELIDES S (2004) Arsenic removal from contaminated water by natural iron ores. Miner. Eng. 17 517-524. http://dx.doi.org/10.1016/j.mineng.2003.11.020 [ Links ]
Received 20 July 2015
Accepted in revised form 3 June 2016
* To whom all correspondence should be addressed.+27 51 401-9656 / +27 62 704 6350; e-mail: erasmuse@ufs.ac.za


{kind=link}
{kind=link}