










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkWater SA
versão On-line ISSN 1816-7950
versão impressa ISSN 0378-4738
Water SA vol.41 no.2 Pretoria 2015
http://dx.doi.org/10.4314/wsa.v41i2.11
Investigation of carbonate dissolution for the separation of magnesium hydroxide and calcium sulphate in a magnesium hydroxide-calcium sulphate mixed sludge
TT RukuniI, *; JP MareeII; FHH CarlssonI
IDepartment of Environmental, Water and Earth Sciences, Tshwane University of Technology, Private Baq X680, Pretoria, 0001, South Africa
IIRand Water Chair in Water Utilisation, Tshwane University of Technoloqy,Private Baq X680, Pretoria, 0001, South Africa
ABSTRACT
South Africa is experiencing a large environmental problem due to uncontrolled discharge of acid mine water into public water courses. The need for neutralisation and desalination of acid mine drainage is a significant issue in South Africa and the sludges that result from mine wastewater treatment usually contain elevated levels of mixed contaminants derived from those originally contained in the wastewater. A more reasonable approach to ultimate sludge disposal is to view the sludge as a resource that can be recycled or reused. Carbon dioxide and a sludge mixture consisting of Mg(OH)2 and CaSO4.2H2O are by-products from acid mine drainage treatment processes. This study was carried out to explore the feasibility of separating Mg(OH)2 from CaSO4.2H2O through dissolution of Mg(OH)2 by accelerated carbonation in a pressurised, completely-mixed reactor. The effects of temperature and pressure, and of both together, on the dissolution of the sludge mixture with time were investigated. Parameters monitored included alkalinity, pH, conductivity and Ca2+, Mg2+ and SO42- concentrations. OLI Analyser Studio Version 9.0 software was used for modelling predictions of chemical speciation of the mixtures. The optimum separation capacity for the Mg(OH)2-CaSO4.2H2O sludge mixture was determined to be 99.34% Mg2+ and 0.05% Ca2+ in the aqueous phase when contacted with CO2 at a temperature of 5°C and pressure of 150 kPa. The model predictions were in agreement with the experimental findings. Temperature and pressure have a significant impact on the dissolution of the mixed sludges when contacted with CO2.
Keywords: Carbonation, gypsum, dissolution, reclamation, carbon dioxide, sludge disposal
INTRODUCTION
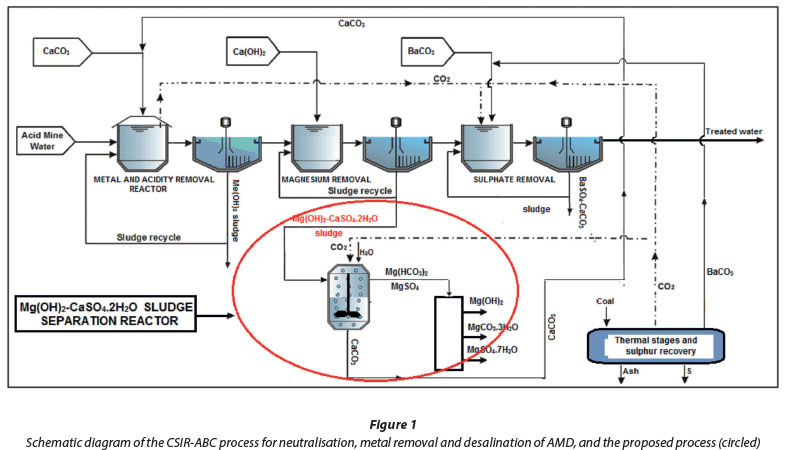
South Africa is experiencing large environmental threats and problems due to uncontrolled discharge of acid mine drainage (AMD) into public water courses. The eMalahleni Water Treatment Works (EWTW) treats 50 Mℓ/d of acid mine water. Limestone is used in the pre-treatment stage for the removal of acid and metals. Partial sulphate removal is achieved through gypsum crystallisation at pH 6. Lime is used for the removal of magnesium as Mg(OH)2 and further sulphate removal is achieved through gypsum crystallisation at pH 11. Ultrafiltration and reverse osmosis are used as a final desalination stage. Separation of Mg(OH)2 from gypsum is needed for the CSIR-ABC process as described by Rukuni et al. (2012a; 2012b). A mixed g(OH)2-CaSO4.2H2O sludge is produced in the second stage of the process. In order to exploit the potential usefulness of the sludge, it is important to separate individual compounds from one another, such as Mg(OH)2 from CaSO4.2H2O. Figure 1 shows a schematic diagram of the CSIR-ABC process. Rukuni et al. (2012a and 2012b) have also studied the separation of CaCO3-BaSO4 and Mg(OH)2-BaSO4 sludge mixtures in contact with CO2.
The aim of this study was to separate magnesium hydroxide from calcium sulphate through dissolution of Mg(OH)2 by accelerated carbonation. By adding CO2 to a Mg(OH)2 + CaSO4.2H2O sludge, Mg(OH)2 dissolution occurs according to the following reaction (Rukuni et al., 2012b):
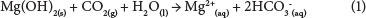
MATERIALS AND METHODS
A sludge mixture of 7.78% CaSO4.2H2O and 92.2% Mg(OH)2 was prepared (mass %). Commercial grade Mg(OH)2 (60 g) and CaSO4-2H2O (5.06 g) were used. The reaction vessel was equipped with a BirCraft stirrer, temperature sensor, pressure gauge and in situ pH and conductivity electrodes. This unit was rated for pressures up to 1000 kPa and a maximum temperature of 150°C.
The main body of the reactor was constructed from a Class 12 unplasticised polyvinylchloride (uPVC) pipe with a wall thickness of 3.2 mm and an internal diameter of 560 mm. The length of the pipe was 520 mm with a uPVC base and top plates that were each 65 mm thick. The dosage points were 300 mm above the effluent take-off point that was fitted at the bottom of the reactor. A pressure regulator was used to control CO2 dosage by monitoring the pressure in the reactor.
Magnesium hydroxide and calcium sulphate dihydrate were added to 1 ℓof deionised water and pumped into the reaction vessel (Watson-Marlow pump) with continuous stirring. The mixed sludge was contacted with CO2 in the 3 ℓcompletely-mixed, pressurised reactor (Fig. 2). Pressurised carbon dioxide was dissolved in water in the pressurised reactor to lower the pH and increase the pressure to desired levels. The particles were kept suspended by a shaft stirrer equipped with a disk-type, radial-flow impeller at the bottom and an axial flow impeller 50 mm above the bottom impeller. Surface carbonation of the suspension occurs from the surface area exposure of the liquid to carbon dioxide and this is enhanced by the vortexing action caused by the axial flow impeller.
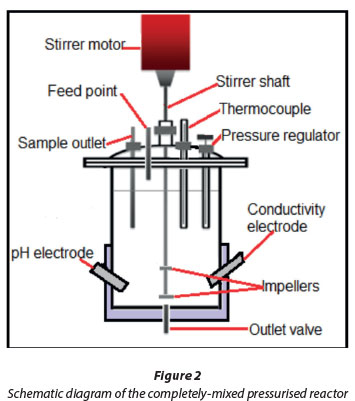
The pH, temperature, electrical conductivity and pressure were measured in situ at predefined sequences of time steps of 0, 10, 20, 40 and 60 min. The predicted and measured effect of the following parameters with time was investigated:
• Temperature: 0, 10, 25, 45°C
• Pressure: 0; 50; 100; 200; 400; 500; 650 kPa
• Mg(OH)2 concentration: 60 g/ℓ
• CaSO4.2H2O concentration: 0, 5.06 g/ℓ
Samples were collected regularly and filtered through a 0.45 μm membrane filter. Sulphate, alkalinity, and pH determinations were carried out manually according to standard procedures (Eaton et al., 1995). Calcium and magnesium were analysed using the EDTA method and atomic absorption spectrophotometry. Alkalinity was determined by titrating the solution to pH 4.3 using 0.1 M HCl. No replicates were analysed.
RESULTS AND DISCUSSION
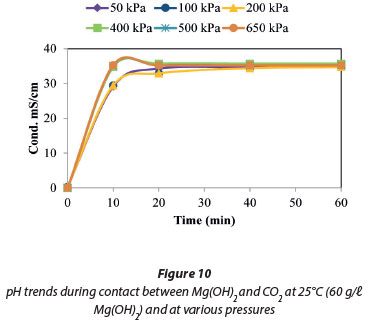
Figures 3 to 20 show the dissolution of Mg(OH)2 (brucite) and CaSO4.2H2O (gypsum) when contacted with CO2 in water under various conditions. Figures 3 to 6 contain the experimental values for the dissolution of gypsum in contact with CO2, while Figs 7 to 11 show the experimental values for the dissolution of Mg(OH)2 in contact with CO2. Figures 12 to 20 show the experimental and predicted findings on the mixed sludge in contact with CO2. Alkalinity (Alk) measurements were used to monitor the formation of Mg(HCO3)2 and Ca(HCO3)2 and include the parameters listed in Eq. (2). As the system became enriched with CO2, the extent of dissolution decreased as a function of changes in the Mg(OH)2 or CaSO4.2H2O saturation state, to yield both magnesium and calcium ions and alkalinity (Rukuni et al., 2012a, Rukuni et al., 2012b).










Dissolution of CaSO4.2H2O in contact with CO2
Effect of temperature
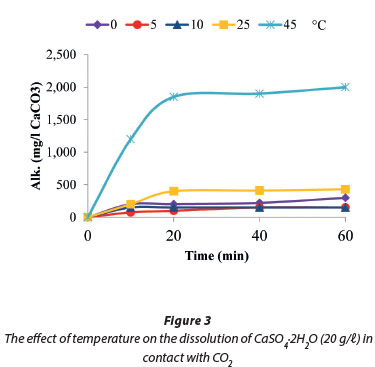
Figure 3 shows that the solubility of gypsum in contact with CO2 decreased with decreasing temperature and vice versa. The highest solubility values of 2 000 mg/ℓ (as CaCO3) were recorded at 45°C compared to 430 mg/ℓ (as CaCO3) at 25°C. Our findings were consistent with those of Al-Khadi et al. (2011) who reported that an increase in temperature from 25°C to 50°C resulted in a corresponding increase in gypsum dissolution, when they investigated the dissolution of gypsum in HCl.
The dissolution of gypsum provided a discernible amount of dissolved calcium and sulphates (151.8 mg/ℓ as CaCO3) in solution in 10 min (Fig. 4) at a temperature of 10°C and was then stable for the remaining 50 min of the reaction time. Figure 4 also shows that the solution pH was stable between 5 and 6. This was attributed to the existence of an unstable layer and fine particle removal from the crystal surface following the transition state theory, while obeying the Power Law (Rukuni et al., 2012b). Raju and Atkinson (1990) and Colombani (2008) reported gypsum solubility in pure water to be: Csat=15 mmol/ℓ.
In the current studies, the calculated solubility of CaSO4-2H2O in carbonated systems at 45°C was 20 mmol/ℓ. This increase in solubility was attributed to the decrease in water activity by addition of CO2 which led to the observed increase in gypsum solubility. The observed decrease in calcium sulphate solubility at temperatures below 45°C was explained by either an over-compensating increase in activity coefficients of calcium and/or sulphate, as reported by Martynowics et al., (1996) or that the pH was not low enough to increase the solubility (Carlberg and Matthews, 1973; Delorey et al., 1996).
Effect of pressure
Figure 5 shows the effect of pressure on the dissolution of gypsum in contact with CO2 and demonstrated that a pressure increase from 50 and 650 kPa had no impact on the dissolution rate at 25°C. Figure 6 illustrates the influence of pressure on pH and electrical conductivity at 50 and 650 kPa and the values were almost the same while following the same trend. The equilibrium conductivity values in the carbonated system increased from 0 mS/cm to 2.2 mS/cm in the initial 10 min of the reaction and then remained constant for the last 50 min, throughout the pressure range. The measured conductivity was in good agreement with the calculated value of k=2.15 mS/cm in pure water (Kuechler et al., 2004).
Dissolution of Mg(OH)2 in contact with CO2
Effect of temperature
The dissolution rate of Mg(OH)2 increased with decreasing temperature as shown in Fig. 7. We also observed that the concentration of Mg2+ in solution increased to almost 36 000 mg/ℓ (as CaCO3) in the first 20 min and then dropped to 22 000 mg/ℓ (as CaCO3) in the last 20 min of the reaction. The maximum solubility value of 36 000 mg/ℓ in the early stages of the reaction could be explained by the high solubility of Mg(HCO3)2, the intermediate product when Mg(OH)2 is contacted with CO2.
The Mg(HCO3)2 will react further with other species in the system to form artinite, brucite, hydromagnesite, magnesite, magnesium carbonate, nesquehonite and periclase (Rukuni et al., 2012b). The Visual Minteq Model was also used in previous studies which reported that some of these compounds exceeded their solubilities as indicated by the positive saturation index values (Rukuni et al., 2012b). The precipitation of these oversaturated compounds would explain the observed drop in Mg(HCO3)2 concentration in solution from 36 000 to 22 000 mg/ℓ Mg(HCO3)2 (as CaCO3).
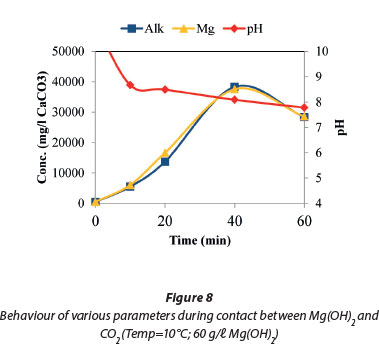
Figure 8 shows the ionic balance between the measured magnesium concentration and the total alkalinity of the system. Over the same period the pH dropped from 9.6 to 7.6 in the first 20 min and stabilised towards the end of the reaction. This confirmed that most of the reaction occurs in the initial 20 min and the further addition of CO2 did not further lower the pH. The dissolution of Mg(OH)2 increased with decreased pH due to the increased formation of soluble Mg(HCO3)2. Therefore, CO2 dosing lowered the pH, and magnesium hydroxide was converted to Mg(HCO3)2.
Effect of pressure
The effect of pressure on the dissolution of Mg(OH)2 was studied at 25°C. The findings plotted in Fig. 9 show that there was no discernible increase in either the dissolution rate or the solution conductivity in the pressure range of 50 to 650 kPa. This was also confirmed by the data plotted in Fig. 10 when the pH and conductivities at 50 and 650 kPa were compared, with no observed changes in the conductivities, pH values or the trends.
Dissolution of a Mg(OH)2-CaSO4·2H2O mixture in contact with CO2
Effect of temperature
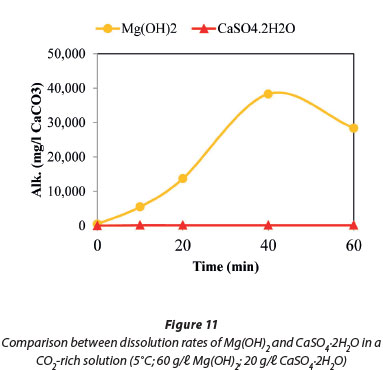
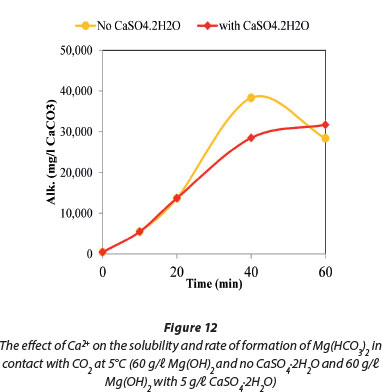
Figure 11 compares the dissolution rates of Mg(OH)2 with CaSO4.2H2O, both in contact with CO2. The results show that the former is more soluble. The dissolution of Mg(OH)2 in contact with CO2 was investigated in the presence and absence of CaSO4.2H2O at 5°C. The data plotted in Fig. 12 show that the presence of CaSO4.2H2O suppressed precipitation of magnesium compounds that resulted in the decrease of Mg(HCO3)2 concentration towards the end of the reaction time.
The mixed Mg(OH)2 and CaSO4.2H2O sludge produced by the CSIR-ABC process cannot be separated by solubility differences due to the low solubility of both Mg(OH)2 (3.8 mg/ℓ Mg(OH)2) and CaSO4.2H2O (2.0 g/ℓ). As CO2 is produced as a waste product in the CSIR-ABC process, it was decided to investigate whether Mg(OH)2 could be separated from the almost sparingly soluble CaSO4.2H2O by dissolving it as Mg(HCO3)2 (Rukuni et al., 2012a; Rukuni et al., 2012b), by contacting the sludge mixture with CO2. The chemical reaction of the sludge mixture with CO2 is shown in Eq. (3) (Bishop, 1943).

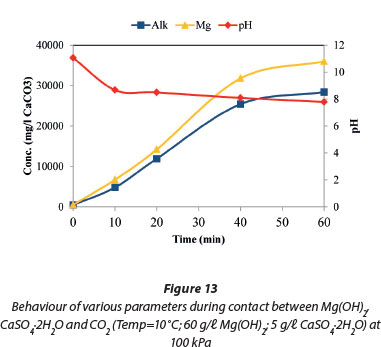
Figure 13 shows that the concentration of Mg2+ ions was 36 000 mg/ℓ (as CaCO3) in the form of MgSO4 and Mg(HCO3)2. This concentration was higher than the aqueous concentration of Mg2+ ions when Mg(OH)2 alone was contacted with CO2, as shown in Fig. 8. This was due to the presence of sulphate ions which suppressed the precipitation of the other magnesium compounds (artinite, brucite, hydromagnesite, magnesite and periclase) except for nesquehonite. This was confirmed by the speciation of the aqueous and solid species, depicted in Figs 17 and 18, respectively.
The total alkalinity of the system increased with decreasing temperature as shown in Fig. 14 and this followed the same trend as in Fig. 7 when Mg(OH)2 alone was contacted with CO2. The only difference between these 2 graphs (Figs 7 and 14) is the absence of the final decrease in temperature in Fig. 7. The absence of Ca2+ in solution was attributed to the formation of CaCO3 and MgSO4 after the dissolution of CaSO4.2H2O.
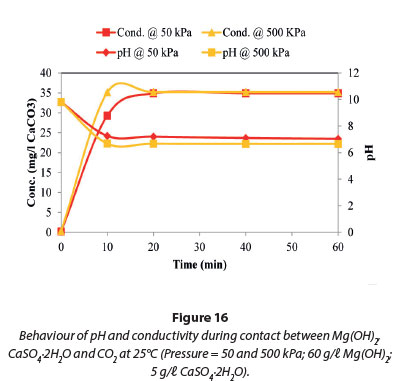
The effect of pressure
The results presented in Figs 15 and 16 show no change in the conductivity and pH when the pressure was varied between 50 and 650 kPa at 25°C. The maximum conductivity in Fig. 15 was comparable to that in Fig. 9, when Mg(OH)2 was investigated. This suggests that the reactions taking place in this system were due to the dissolution of Mg(OH)2 as Mg(HCO3)2.
Based on model calculations using OLI Analyzer Studio Version 9.0 software, Figs 17 and 18 show the effects of pressure on the dissolution of Mg(OH)2 and CaSO4.2H2O in contact with CO2 at 25°C. The results show that most of the Mg would be in solution in the form of MgOH+ and MgHCO3+ while nearly all of the dissolved calcium would be precipitated as CaCO3 (Fig. 18). The amount of Mg species in solution increased linearly between 1 and 650 kPa and will be constant (110 000 MgOH+ and 90 000 mg/ℓ MgHCO3+) from 800 to 1 500 kPa.
Although most of the calcium was precipitated as CaCO3 and CaSO4, nesquehonite (MgCO3.3H2O) constituted the larger percentage of the solids at pressures between 101 and 304 kPa. All nesquehonite will be in solution at pressures above 300 kPa and the solids will be mainly CaSO4 and CaCO3. The CaSO4 concentration also dropped with increasing pressure to zero at 800 kPa. The drop in MgCO3-3H2O and CaSO4 content in the solid phase may lead to the formation of CaMg(SO4)2 as shown in the reaction (Eq. (4)):

The amount of CaCO3 in solution remained almost constant throughout the pressure range.
The separation of Mg(OH)2 and CaSO4.2H2O
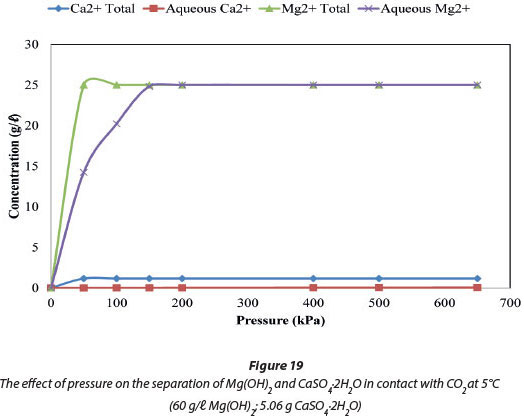
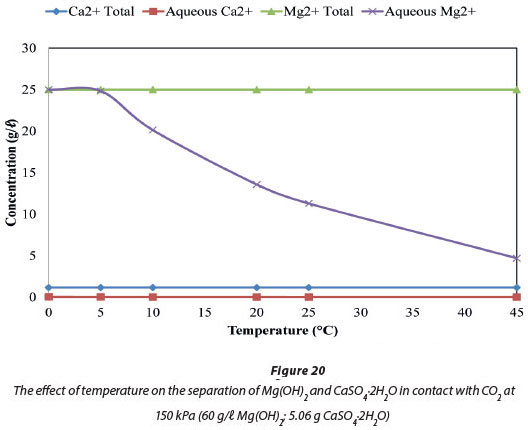
Figure 19 compares the total amount of Mg and Ca ions in the system, by mass, to the amount of these ions in the aqueous phase at a fixed temperature of 5°C. Figure 20 compares the ions at a fixed pressure of 150 kPa. Figure 19 shows that 99.34% of the system's 25 005 mg/ℓ Mg was in solution at 150 kPa when the temperature was maintained at 5°C, and all of the Mg was in solution at pressures from 2 to 650 kPa. Figure 20 shows that when pressure is fixed at 150 kPa, 100% (25 005 mg/ℓ Mg) of the dosed Mg ions were in solution at 0°C, and 99.34% (24 840 mg/ℓ Mg) were in solution at 5°C. In the case of Ca ions, 1.14% by mass of the total 1 164 mg/ℓ Ca was dissolved and will be the major impurity in the dissolved Mg(HCO)3. The Ca2+ impurity in the recovered Mg2+ constitutes 0.05% by mass.
These results suggest that the dissolution of calcium sulphate in carbonate systems occurs in the following manner:

The calcium sulphate also reacts with bicarbonate ions to form CaCO3 and HSO4-. The bisulphate formed will then liberate H+ (Eqs (12) and (14)), which will lower the solution pH and then promote further dissolution of CaSO4.2H2O:
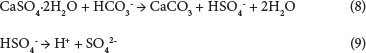
From the equations above, the solubility of gypsum increases because of an increase in ionic species in solution, thus increasing the interaction between the reacting species that elevates the solubility limit to the formation of both CaCO3 and Ca(HCO3)2The initial dissolution stage of gypsum is attributed to the removal of an unstable layer and fine particles from the crystal surface following the transition state theory while obeying the power law:

Where Rdiss. is the dissolution rate, ks the surface reaction rate constant, Csis the concentration of the dissolved species at the surface, Csat is the solubility and n is a constant (Lasaga, 1998).
As the system becomes enriched in CO2, the dissolution rate of Mg(OH)2 could be influenced by its saturation level, Ω (Rukuni et al., 2012b):

Where Ksp is the solubility product; T is temperature; S is solubility and p is the pressure (Mucci, 1983).
Note: All the square-bracketed species are stoichiometric concentrations (molality or moℓ/kg) and disregard any complex formation or ion pairs. It is thus necessary to specify not only the pH scale used in the evaluation of the dissociation constants (Dickson, 1984), but also their dependency on ionic strength, temperature, and pressure.
During the dissolution of CaSO4.2H2O, a mass transport boundary layer develops in the vicinity of the solid particles and dissolution will then proceed in a manner whereby the ions are firstly unbound from the solid and solvated. The second step is when the ions migrate through the diffusion boundary layer. The concentration is generally considered as linear in this layer and Fick's Law follows:

Where Rdiftis the diffusion rate, kt = D/δa transport rate constant, D the diffusion coefficient of the dissolved components, δ the diffusion boundary layer thickness and c is the concentration in the bulk liquid (Lasaga, 1998). The final step is where the ions are advected throughout the solution. These steps are shown in Fig. 21.
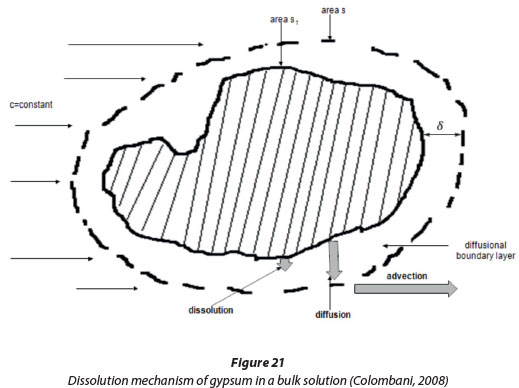
Sjöberg and Rickard (1984) studied the dissolution of calcite and gypsum and found that the dissolution of carbonate and sulphate minerals exhibit mixed kinetics where the kinetics are due to both diffusive transport and controlled by chemical reaction. When Rdift<< Rdiss, the phenomenon of the dissolution kinetics is transport controlled, and when Rdift»Rdiss, the kinetics are reaction based.
Liu and Nancollas (1971) noticed that the rate of dissolution is proportional to the difference between the instantaneous concentration C, at time t, and the gypsum saturation solubility in water, CS. This statement can be formulated mathematically as follows:

The value of the saturation concentration of the calcium ions in water can be derived by knowing that 2.5 kg of gypsum are dissolved in 1 m3 of water. Since the molar mass of gypsum is 172.2 g/mol, the saturation concentration in 1 m3 of water is equal to 14.52 mol/m3 (Betti et al., 2008).
Building upon the findings of previous growth and dissolution studies of other mixed sludges, it was concluded that the dissolution rates of these minerals are governed by solvation affinity for the near-surface divalent metals. The results of this study suggest that the dissolution kinetics of sulphates is governed by a similar, but inverse, rate-limiting step. That is, dissolution rates are proportional to the hydration or solvation affinity of metals contained in these structures. In gypsum, the divalent atoms of Ca are coordinated to join with the SO4 tet-rahedra. Rates of gypsum dissolution are also quite rapid and rates are limited by transport control in most conditions (Liu and Nancollas, 1971; Barton and Wilde, 1971). However, initial rates of dissolution appear to be surface reaction controlled, as suggested, by a relatively large activation energy of 40 kJ/ mol (Liu and Nancollas, 1971).
The separation of Mg(OH)2 and CaSO4.2H2O from a Mg(OH)2-CaSO4.2H2O mixed sludge was shown to be feasible by accelerated carbonation. However, it was also noted that CaSO4.2H2O will not be recovered as CaSO4, but the calcium will be recovered as CaCO3 while Mg(OH)2 will be recovered as either MgCO3.3H2O, Mg(OH)2 or MgSO4.7H2O.
MgCO3-3H2O and Mg(OH)2 can be precipitated from MgHCO3+ and MgOH+ by lowering the reaction pressure to atmospheric. The MgCO3.3H2O and Mg(OH)2 can still be reused in the CSIR-ABC process for the neutralisation of free acid and metal removal during the treatment of acid mine drainage.
The practical, optimal operating conditions for the dissolution process are thus temperatures close to 0°C and pressures as close as possible to atmospheric (50 to 200 kPa), depending on the application. From the reaction equilibrium and the results of this study, it was apparent that decreasing the temperature and lowering CO2 pressures will result in an improved conversion of Mg(OH)2 to Mg(HCO3)2. Keeping the pressure low will help in lowering the solubility of the CaCO3, hence increasing the separation rate.
Based on our current application, the recommended optimum operational conditions of temperature and pressure on the separation of Mg(OH)2 and CaSO4.2H2O in contact with CO2 is a temperature of 5°C and pressure of 150 kPa, to yield a 99.34% Mg2+ recovery as Mg(HCO3)2 with 0.05% Ca2+ impurities as Ca(HCO3)2.
Applying the results of this study will contribute to the improvement of the CSIR-ABC process, designed to meet the criteria for maximum value of treated water and by-products, coupled with lowered running and sludge disposal costs. The cost of the separation process is low because all the process raw materials (CO2 and the Mg(OH)2-CaSO4.2H2O sludge) are waste products of the CSIR-ABC process (Fig. 1). It is also foreseen that the cost can be kept low in other applications by producing CO2 on site by burning coal and scrubbing the off-gases in water rather than purchasing pure CO2.
CONCLUSIONS
Carbon dioxide can be used for separation of Mg(OH)2 from CaSO4.2H2O due to the high solubility of Mg(HCO3)2 compared to CaSO4-2H2O. The optimum separation capacity for the Mg(OH)2-CaSO4.2H2O sludge mixture is 99.34% Mg2+ and 0.05% Ca2+ in the aqueous phase when contacted with CO2 at a temperature of 5°C and pressure of 150 kPa.
The model predictions were in agreement with the experimental findings. Temperature and pressure had a significant impact on the dissolution of the mixed sludges when contacted with CO2.
ACKNOWLEDGMENTS
Financial support from The National Research Foundation's Technology and Human Resources for Industry Program (THRIP) and the use of the laboratory facilities at Tshwane University of Technology are gratefully acknowledged.
REFERENCES
AL-KHADI MH, AL-JUHANI AM, AL-MUTAIRI SH and GURMEN MN (2011) New insights into the removal of calcium sulphate scale. Saudi Aramco J. Technol. 1 14-51. [ Links ]
BARTON AFM and WILDE NM (1971) Dissolution rates of polycrystalline samples of gypsum and orthorhombic forms of calcium sulphate by a rotating disc method. Trans. Faraday Soc. 67 3590-3597. [ Links ]
BETTI D, BUSCARNERA G, CASTELLANZA R and NOVA R (2008) Numerical analysis of the life-time of an abandoned gypsum mine. In: The 12th International Conference of International Association for Computer Methods and Advances in Geomechanics (IACMAG) 1-6 October 2008, Milan, Italy. [ Links ]
BISHOP DL (1943) Function of carbon dioxide in producing efflorescence on plaster and cement products. J. Res. Natl Bur. Stand. 30 361-366. [ Links ]
CARLBERG BL and MATTHEWS RR (1973) Solubility of calcium sulphate in brine. In: SPE Oil Field Chemistry Symposium, 24-25 May 1973, Denver, Colorado, USA. [ Links ]
COLOMBANI J (2008) Measurement of the pure dissolution rate constant of a mineral in water. Geochim. Cosmochim. Acta 72 5634-5640. [ Links ]
DELOREY J, ALLEN S and MCMASTER L (1996) Precipitation of calcium sulphate during carbonate acidizing: Minimizing the risk. In: Petroleum Society of Canada 47th Annual Technical Meeting, 10-12 June 1996, Calgary, Alberta, Canada. [ Links ]
DICKSON AG (1984) pH scales and proton-transfer reactions in saline media such as seawater. Geochim. Cosmochim. Acta 48 2299-2308. [ Links ]
EATON AD, CLESCERI LS and GREENBERG AE (eds) (1995) Standard Methods for the Examination of Water and Wastewater. APHA, AWWA and WEF, USA. [ Links ]
KUECHLER R, NOACK K and ZORN T (2004) Investigation of gypsum dissolution under saturated and unsaturated water conditions. Ecol. Modell. 176 1-14. [ Links ]
LASAGA AC (1998) Kinetic theory in the earth sciences, Princeton University Press Princeton, New Jersey, USA. [ Links ]
LIU ST and NANCOLLAS GH (1971) The kinetics of dissolution of calcium sulphate di-hydrate. J. Inorg. Nuclear Chem. 33 2311-2316. [ Links ]
MARTYNOWICS ETMJ, WITKAMP GJ and VAN ROSMALEN GM (1996) The effect of aluminum fluoride on the formation of calcium sulfate hydrates. Hydrometallurgy 41 171-186. [ Links ]
MUCCI A (1983) The solubility of calcite and aragonite in seawater at various salinities, temperatures and 1 atmosphere total pressure. Am. J. Sci. 283 780-799. [ Links ]
RAJU K and ATKINSON G (1990) The thermodynamics of ''scale" mineral solubilities. 3. Calcium sulfate in aqueous NaCl. Chem. Eng. Data 35 361-367. [ Links ]
RUKUNI TT, MAREE JP and ZVINOWANDA CM (2012a) Recovery of calcium carbonate from a calcium carbonate-barium sulphate mixed sludge. In: 27thInternational Conference on Solid Wastes and Technology, 11-14 March 2012, Philadelphia, USA. [ Links ]
RUKUNI TT, MAREE JP and ZVINOWANDA CM (2012b) Separation of magnesium hydroxide and barium sulphate from abarium sulphate - magnesium hydroxide mixed sludge by carbonation: The effect of temperature. J. Civ. Environ. Eng. 2 116. doi:10.4172/2165-784X.1000116. [ Links ]
SJÖBERG EL and RICKARD DT (1984) Calcite dissolution kinetics: surface speciation and the origin of the variable pH dependence. Chem. Geol. 42 119-136. [ Links ]
* To whom all correspondence should be addressed. e-mail: RukuniTT@tut.ac.za or tece.1997@yahoo.com


{kind=link}