










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkWater SA
versión On-line ISSN 1816-7950
versión impresa ISSN 0378-4738
Water SA vol.40 no.3 Pretoria jun. 2014
Determination of naproxen, ibuprofen and triclosan in wastewater using the polar organic chemical integrative sampler (POCIS): A laboratory calibration and field application
Robert Amdany; Luke Chimuka; Ewa Cukrowska
Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, P/Bag 3, Braamfontein, Johannesburg 2050, South Africa
ABSTRACT
In this study, the occurrence in wastewater of two non-steroidal anti-inflammatory drugs (NSAIDs), naproxen and ibuprofen, and one personal care product, triclosan, was assessed using the polar organic chemical integrative sampler (POCIS). The samplers were initially calibrated in the laboratory to obtain sampling rates (Rs ) for each target compound followed by deployment in the influent and effluent of Goudkoppies and Northern Wastewater Treatment Plants (WWTPs), South Africa. Exposure was done for 14 days in 2012. High performance liquid chromatography (HPLC) system with ultraviolet (UV) and fluorescence (FLD) detectors was used to analyse POCIS extracts. Laboratory calibration of POCIS yielded R values for the three compounds that were between 0.087 and 0.383 ℓ-d-1 in quiescent conditions, and 0.125 and 0.936 ℓ-d-1 in stirred conditions. From the accumulated amounts in field-deployed samplers, estimated freely dissolved concentrations of the studied compounds in wastewater influent ranged from 55.0 to 78.4 µg-ℓ-1 and 52.3 to 127.7 µg-ℓ-1 in Goudkoppies and Northern WWTPs, respectively. Average concentrations of these compounds in the treated effluent ranged from 10.7 to 13.5 µg-ℓ-1 in Goudkoppies WWTP, and 20.4 to 24.6 µg-ℓ-1 in Northern WWTP. Analyte removal efficiencies varied between 68 and 86% in Goudkoppies WWTP and 61 and 82% in Northern WWTP. Grab samples processed by SPE method yielded higher analyte concentrations (up to three-fold) as compared to POCIS-derived estimates. This discrepancy was attributed to SPE's ability to extract both the free dissolved, and particle sorbed fractions of the contaminants.
Keywords: Polar organic chemical integrative sampler, pharmaceuticals and personal care products, wastewater, wastewater treatment plants
INTRODUCTION
Pharmaceuticals and personal care products (PPCPs) comprise an emerging and very diverse group of polar organic contaminants characterised by active functional groups designed to elicit certain physiological responses in target organisms. PPCPs are a group of organic micropollutants whose presence in the environment has aroused intense attention among environmental scientists and the general public due to their known and/or perceived health effects on animals and humans. They principally enter the aquatic environment through treated effluent discharged from WWTPs. As a result of their incomplete degradation in the treatment plants, many of these chemicals survive the elimination process and some are even returned to their biologically active forms through de-conjugation (Braga et al., 2005; Miao et al., 2004). The existence of PPCPs at trace and ultra-trace levels in water systems, coupled with frequent temporal variations, render most traditional sampling approaches insufficient. Passive sampling devices (PSDs) have been proposed as suitable alternatives because they usually combine sampling, selective analyte isolation, pre-concentration and, in some cases, speciation preservation in a single step (Vrana et al., 2005). PSDs provide time-weighted average (TWA) values that take into account fluctuations in pollutant concentrations, target the water-dissolved fraction of the contaminants, and facilitate detection of very low analyte concentrations due to their elongated exposure periods (Kot et al., 2000; Sabaliunas, and Sodergren, 1997). Currently, the polar organic chemical integrated sampler (POCIS) has emerged as the best sample preparation technique for PPCPs (Morin et al., 2012; Alvarez et al., 2004; Jones-Lepp et al., 2004). The device comprises a solid sorbent-receiving phase sandwiched between 2 microporous polyethersulphone (PES) diffusion-limiting membranes firmly held in place by 2 compression rings made of steel or plastic (Alvarez et al., 2007). The sampler's suitability encompasses a variety of hydrophilic organics such as pharmaceuticals, personal care products and pesticides with octanol-water partition coefficients (log Kow ) in the range of 3.0 and 4.0.
The current study was aimed at evaluating the use of POCIS for the assessment of naproxen, ibuprofen and triclosan in wastewater. Determination of the sampling rates (Rs) for the target compounds in the POCIS was undertaken under laboratory conditions and the resultant Rs values applied to field deployments of the samplers at 2 WWTPs serving Johannesburg city, South Africa. A comparison of the POCIS-derived analyte concentrations with values obtained from grab samples from the same sites as processed using solid phase extraction was also done.
EXPERIMENTAL
Chemicals and reagents
Naproxen and ibuprofen (all > 98% pure) were purchased from Sigma Aldrich GmbH (Steinheim, Germany). Triclosan (> 97% pure) was sourced from Merck (Darmstadt, Germany). HPLC-grade methanol (MeOH), and acetonitrile (ACN) were purchased from Sigma Aldrich (Johannesburg, South Africa) and B & J brand (Honeywell, Germany). Formic acid, citric acid and disodium hydrogen phosphate (all > 97% pure) were obtained from Sigma Aldrich (Johannesburg, South Africa). PES membranes and Oasis HLB sorbent were purchased from Waters Corporation (Milford, MA, USA). Standard POCIS steel rings were fabricated at the Mechanical Engineering workshop (University of the Witwatersrand, South Africa). Deionised water was obtained from a Millipore Simplicity 185 system (Millipore, Bedford, MA, USA). All other reagents used were of the highest analytical grade.
Stock solutions were constituted in ACN at a concentration of 1 000 µg-mℓ-1 and working solutions (standards and calibration solutions) prepared from the stock solutions in ACN and used in the development and validation of the analytical methods.
POCIS assembly and deployment
200 mg of pre-cleaned Oasis HLB sorbent was weighed and superimposed between two 90 mm diameter PES membranes and held in place with two stainless steel compression rings to safeguard against sorbent losses. Bolts and screws were used to tighten the rings. Prepared POCIS devices were then stored in the fridge at 4°C awaiting deployment. At the sampling sites, POCIS devices were unpacked from the transport container, mounted onto the deployment device, and quickly immersed into the wastewater at a depth of between 1 to 1.5 m and anchored using a polyethene rope to keep them in the desired position. Each deployment was done in triplicate. Atmospheric contribution of the contaminants at each sampling site was assessed using field controls. After the exercise, the controls were transported to the laboratory and refrigerated at 4°C awaiting retrieval and eventual processing of the deployed POCIS.
Sampling sites
Figure 1 shows a map of the sampling sites: Goudkoppies and Northern WWTPs.

Goudkoppies WWTP
Goudkoppies WWTP is located in Marshalltown, about 15 km south of Johannesburg city central business district (CBD). It treats domestic, industrial and hospital effluent discharged from Johannesburg CBD and the residential areas situated to the south-east of the city. It also receives sewage discharges from several clinics and hospitals. Treated effluent from this WWTP is eventually discharged into the Klip River through a small stream bordering the plant. The facility was commissioned in 1978 and routinely treats between 120 and 150 mill. ℓ.day-1. It also produces about 15 804 t dry sludge annually. The plant consists of a new head of works with screening, degritting, primary sedimentation, raw sludge thickening/acid fermentation, flow balancing, activated sludge incorporating the 5-stage phoredox process, final clarification, chlorination, waste sludge thickening, digestion, dewatering and solar drying of sludge (Johannesburg Water, 2012). Interested farmers collect the dried sludge and use them on their privately owned farmlands.
Northern Works treatment plant
Northern Wastewater Treatment Works is the biggest WWTP in Johannesburg. The treatment plant located at Diepsloot, about 38 km north of Johannesburg CBD, was designed to handle up to 360 mill. ℓ.day1 of raw effluent and incorporates a 90 t.day-1 composting unit as well. However the average flows treated daily stand at 323 mill. ℓ, and 28 392 t of dry sludge is produced annually by the plant (Johannesburg Water, 2012). The treatment works mainly collects and treats household sewage from many residential areas including those situated in the northern parts of Johannesburg. Treated effluent is ultimately discharged into the Jukskei River. The facility consists of 4 units that in total perform the following activities: degritting, lime addition, primary sedimentation, primary and secondary biological filtration, primary humus removal, acid fermentation of raw sludge, flow balancing, activated sludge bioreacting, chlorination, sludge thickening, dewatering and composting (Johannesburg Water, 2012).
Sampler retrieval
The deployment devices were pulled out of the wastewater after severing the cable ties. Samplers were then unscrewed from the deployment cages, and, where applicable, biofouling removed from the surfaces of the PES membranes. Thereafter, the devices were placed in air-tight, clean metal cans, and transported to the laboratory where storage was done at -20°C until processing.
Processing
POCIS devices were carefully disassembled and the sorbent transferred into 3 ml empty SPE cartridges using a few drops of ultrapure water. Packing of the cartridges was done under vacuum using a SPE Manifold and to safeguard against losses, polyethylene frits were placed before and after packing sorbents in the cartridges. The sorbent was dried under mild vacuum until a constant weight was obtained. Where elution was not possible immediately, cartridges were refrigerated at 4°C. After adding recovery standards, analytes were eluted with 2 portions of 3-mℓ volumes of eluents: 1 x 3 mℓ of 100% acetonitrile (ACN) followed by 1 x 3 mℓ of ACN:MeOH, 50:50 (v/v). Sample dilution was performed where necessary. Samples were finally transferred into a 2-mℓ GC-vial and analysed subject to instrumental analysis.
POCIS calibration
In this study, the calibration of POCIS was done based on the methods developed by Macleod et al. (2007) and Li et al. (2010). Modifications of certain aspects of the method were, however, made during the optimisation process. As shown in Eq. (1), information on the decrease in analyte concentration in the bulk solution (Cw) over time is used to determine the sampling rate, Rs .

where:
kU and kD are uptake and dissipation rate constants in ℓ.d-1, respectively
Cw(0) and Cw(t) are the initial water concentration (time = 0) and concentrations at time t in µg.ℓ-1, respectively
Equation (2) arises from the rearrangement of Eq. (1):

When (Ln[Cw(t)/Cw(0)]) is plotted against exposure time, its slope gives a close estimate of the k value. The corrected uptake rate constant for the target compound can be obtained by subtracting the dissipation rate constant from the average rate constant, k.
Sampling rate can thus be determined using Eq. (3):

where:
VT is the total volume of the bulk water being extracted
The experimental set-ups involved static exposure of POCIS to target compounds for 7 and 14 days. A 2 ml mixture containing 15 µg of naproxen, ibuprofen and triclosan was added to the bottom of a clean 3 ℓ capacity beaker and the constituting solvent (ACN) evaporated completely. Ultrapure water (3 ℓ) was added to the beaker and analytes left to equilibrate for about 2 h resulting in a nominal concentration of 5 µg ℓ-1 of each compound. Deployment of POCIS in the solution was preceded by initially soaking them in ultrapure water for about 12 h so as to reduce the possibility of sudden increase in flux across the membrane during the wetting stage at the beginning of the experiment (Mazzella et al., 2007). A single sampler was then suspended vertically in the solution for the exposure period at room temperature (25°C) and the beaker covered with aluminium foil in order to limit exposure to light and to minimise losses arising from volatilisation. Experiments were conducted under both stirred and quiescent conditions to investigate the influence of turbulence on uptake of analytes. For stirred conditions, agitation of the analyte-fortified solution was done at 400 r∙min-1 using a C-MAG HS 10 magnetic stirrer (Staufen, Germany). In the quiescent arrangement, the solution was slowly agitated at the bare minimum speed (60 r∙min-1) of the magnetic stirrer to prevent development of a concentration gradient in the vessel. Parallel experiments were conducted using solutions with analyte nominal concentrations of 10 µg∙ℓ-1. Decrease in water concentration over time was monitored by excising 20 ml aliquots of the bulk solution after every 24 h, processing by solid phase extraction (SPE), and subsequently analysing with HPLC-UV/FLD. Since dissipation of target compounds in the positive blanks was not evident in this study, kU was assumed to be equal to k and, hence, sampling rates, Rs, for individual compounds were then calculated using the derived kU value and 3 ℓ as the volume of the water used (Eq. (3)). Positive control experiments, i.e., those containing only fortified water without the POCIS, were carried out in order to account for possible analyte degradation, sorption or volatilisation during exposure. Negative controls in which the POCIS was exposed to reagent water devoid of the analytes, and aimed at checking for analyte contamination, were done. All experiments ran concurrently. The quality fit for the regressions was characterised by the correlation coefficient, and consistency was evaluated based on the standard deviations of the kU values from triplicate experiments.
Solid phase extraction (SPE)
The 20 ml aliquots from the laboratory experiments and 150 ml aliquots of environmental samples (influent and effluent) from the WWTPs were extracted by solid phase extraction (SPE). Initially, wastewater samples were filtered through 1.0 µm mesh size glass fibre filters. Oasis HLB cartridges were conditioned with 3 mℓ ethyl acetate, 3 mℓ acetonitrile (ACN), and 3 mℓ of ultrapure water, sequentially, ensuring that the cartridge did not dry out between loadings. All samples were adjusted to pH 4 using 1 M sulphuric acid solution. With the aid of a vacuum pump (Gilson, Williers Le Bel, France), 150 mℓ of the field samples were then passed through the cartridge at 3 mℓ-min-1. Washing was done using 3 mℓ of ACN:H2O (5:95, v/v) followed by 3 mℓ of n-hexane at a flow rate of 1 mℓ-min-1. Thereafter, the cartridges were dried under mild vacuum for 20 min. Elution was effected sequentially with 1 x 3 ml ACN followed by 3 ml formic acid (2%) in ACN. Finally, the extracts were evaporated to dryness and then reconstituted in 0.5 ml ACN. Procedural blanks were extracted concurrently with the water and wastewater samples.
Instrumentation
A Waters 530 (LA, California, USA) HPLC system equipped with a fluorescence detector was used for the analysis of the pharmaceuticals while triclosan was analysed with a Metrohm Bischoff AG HPLC system (Herisau, Switzerland) equipped with a UV detector. Separation was achieved using a Phenomenex C reversed-phase column (Supelco, Bellefonte, PA, USA) with the dimensions: 250 mm x 4 mm, 5 µm i.d., and carried out in isocratic mode at a flow rate of 0.8 mℓ-min-1 with the mobile phase composing ACN:H2O (0.2% formic acid), 70:30 v/v. The mobile phase was first degassed offline using a Transsonic 460 ultrasonic bath (Elma, Singen, Germany) for 15 min and the injection volume set at 20µℓ The photochemical behaviours of naproxen and ibuprofen were investigated using a RF-10xL fluorescence detector (Shimadzu corporation, Japan) with the excitation/emission wavelengths set at 333/540 (Naproxen) and 220/400 (ibuprofen), while triclosan was detected using a Bischoff Lambda 1010 DAD UV detector (Leonberg, Germany) at an optimum wavelength of 280 nm.
Quality control
Fabrication blanks
200 mg of clean sorbents were transferred into 3 ml empty SPE tubes and secured with PE frits. Three replicates were prepared and used as blank laboratory controls. The cartridges were stored at 4°C until the simultaneous elution and analysis with the POCIS extracts.
Field blank
Another triplicate set of POCIS, each with 200 mg of clean sorbent, were prepared and used as field controls. These controls were transported to the sample site during sampler deployment and then brought back to the laboratory after exposure to atmospheric conditions at the sampling site. Once in the laboratory, they were stored at 4°C. They were simultaneously processed with field exposed samplers.
RESULTS AND DISCUSSION
Analytical performance and quality assurance
During calibration, no target analytes were detected in aliquots of water or in POCIS extracts from the negative blank control. Likewise, POCIS extracts from laboratory procedures and the field blanks yielded no detectable amounts of target compounds. Moreover, no observable dissipation of the analytes was recorded in the positive controls. Thus, blank corrections were not necessary. Solid phase extraction (SPE) of the analytes yielded recoveries that ranged from 75% to 108%. Mean recoveries for individual compounds were as follows: naproxen (92%), ibuprofen (108%), and triclosan (75%). Standard deviations between replicate measurements were all less than 10%. Table 1 presents detection limits (LODs), quantification limits (LOQs) and linearity figures. LOD and LOQ values were defined as the analyte concentrations that produced a peak with a signal-to-noise ratio of 3 and 10, respectively, and the lowest calibration standard solution for each target analyte was used in their determination. LODs and LOQs for the analytes ranged from 0.2 to 4.1 µg.ℓ-1, and from 0.7 to 17 µg.ℓ-1, respectively. All of the compounds exhibited good linearity with coefficients of determination (r2) higher than 0.9982.

Mass balance
Mass balances for the PPCPs were calculated using Eq. (4) below (Macleod et al., 2007).

where:
MA = [the amount extracted by POCIS + the sum of the mass excised during sampling + the mass in water at the end of the uptake experiment + the mass dissipated as measured from the control tank]
MD = mass of analyte before POCIS extraction.
The calculated mass balances for the PPCPs were as listed: naproxen (89%), ibuprofen (97%), and triclosan (64%). These values were comparable to those reported by Li et al. (2010). It was observed that the personal care product, triclosan, gave a lower mass balance as compared to the pharmaceuticals. This may be resulting from degradation of the compound after sequestration by the sorbent, or thereafter. Indeed, Macleod et al. (2007) also reported low mass balance for triclosan and they attributed this to the instability of the compound and/or poor recovery during POCIS extraction.
Uptake kinetics of the PPCPs and sampling rate
PPCP uptake by POCIS can be determined based on the decline in water concentrations over time (Macleod et al., 2007) or the amounts of analytes accumulated in the device over the exposure period (Mazzella et al., 2007; Togola and Budzinski, 2007). In the present study, Rs of the target compounds was calculated based on the decrease in their concentrations over time, i.e., Ln[Cw(t)/ Cw(0)] (Eq. (2)). Analyte loss from the fortified water was linear over the entire 7-day exposure period. Figure 2 presents a plot of LnCw(t)/Cw(0) against exposure time used to determine PPCP loss due to POCIS uptake over 7 days of exposure at 25°C. Good correlation coefficients of the linear regressions were obtained with r2 for individual compounds as follows: naproxen (0.994), ibuprofen (0.998) and triclosan (0.987).

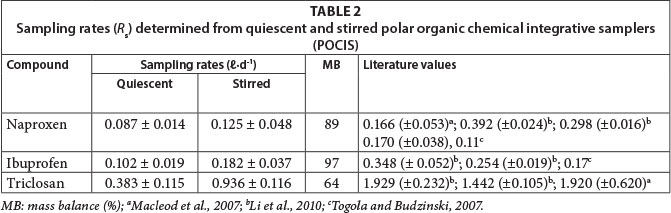
Estimated uptake rate constants obtained from these plots showed good precision as indicated by their standard deviations for triplicate determinations. Table 2 presents the Rs values of the compounds determined at 25°C under both quiescent and stirred conditions.
Analyte Rs values ranged from 0.087 to 0.983 l-d-1 in quiescent experiments, while stirred conditions yielded values that varied between 0.125 and 0.925 ℓ.d-1. These figures were broadly consistent with those reported by other workers (Li et al., 2010; Macleod et al., 2007; Togola and Budzinski, 2007) in similar calibration set-ups.
The influence of hydrodynamics on Rs was visible when values derived from quiescent water were compared to those obtained under stirred conditions. Stirring gave higher analyte sampling rates than quiescent set-ups, and for all study compounds a less than 3-fold increase in the Rs was observed between the two conditions. This is an indication that in POCIS the water boundary layer may be controlling the rate of mass transfer (Alvarez et al., 2007, 2004; Mazzela et al., 2007; Togola and Budzinski, 2007). The influence of flow velocities on accumulation kinetics of hydrophobic compounds by SPMDs has been reported by Vrana and Schuurmann (2002). SPMD sampling rates are affected by environmental exposure conditions with differences as great as 10-fold for flow effects and 4-fold for temperature effects (Li et al., 2010). Comparatively, it seems the hydrodynamic effects on R are not as pronounced in POCIS as in SPMDs.
Field deployment of pharmaceutical POCIS
Occurrence of PPCPs in POCIS extracts
Laboratory-derived Rs were applied in the estimation of TWA concentrations from the amounts of the analytes sequestered by POCIS. The samplers were deployed for 14 days at 2 WWTPs located around Johannesburg city. The deployment exercise was done in August and September 2012. Average water temperatures at both treatment plants remained relatively stable (20 ± 20C) over the entire deployment period. Mean pH values at Goudkoppies and Northern WWTPs were between 7.9 and 8.2 and 8.1 and 8.4, respectively, during exposure. Insofar as no analytes were detected in the field blank extracts, corrections were not made for them. Estimated concentrations (µg.ℓ-1) of the target analytes in both the treated effluent and influent are presented in Table 3, while Fig. 3 shows sample chromatograms of the POCIS extract.
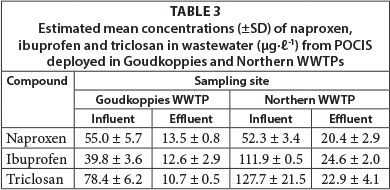
PPCP concentrations in the influent of Goudkoppies WWTP ranged from 39.8 to 78.4 µg.ℓ-1 and 10.6 to 13.5 µg.ℓ-1 in the treated effluent. Northern WWTP yielded compound residues which ranged in concentration from 52.3 to 127.7 µg.ℓ-1 (influent) and 20.42 to 24.58 µg.ℓ-1 (effluent). Expectedly, effluent levels were lower than influent ones, courtesy of degradation activities experienced in the treatment process.
Comparatively, PPCP concentrations in the influent and effluent of Northern WWTP were significantly higher than those detected in Goudkoppies WWTP. While the differences in influent analyte levels may be ascribed to the population served by the WWTP, those in the treated effluent depend on the removal efficiency of the individual facility. In essence, Northern WWTP serves a relatively large area and receives domestic sewage from a population of close to 1 mill. people. In addition, many other diffuse sources, such as discharges from several hotels and restaurants, hospitals and clinics, and industries, are adding to the contaminant load. On the other hand, Goudkoppies WWTP serves a good portion of the city centre, as well as some residential areas located in south-eastern Johannesburg. Its catchment area is smaller than that of the Northern treatment facility. Part of its sewage is of industrial origin with household, hospital and hotel sources accounting for the remainder.
SPE versus POCIS estimates
Field application of the sampler was evaluated by comparing the POCIS-derived data with those from SPE extraction of grab samples. Concentrations from triplicate sets of grab sample analyses obtained during sampler deployment and retrieval were averaged (n = 6) and compared with TWA concentrations from POCIS (Figs. 4 and 5). In spite of the SPE method giving relatively higher values, analyte levels from both techniques were generally comparable.

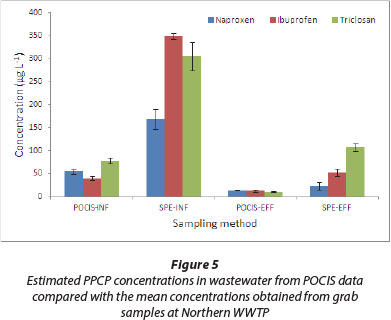
It therefore implies that our laboratory-derived Rs values were within reasonable limits and, hence, appropriate for determination of TWA concentrations. Even though complete characterisation of temporal trends in analyte concentrations is not possible with grab sampling, this approach, however, provides a general picture of the concentration range in the study site. Obtaining TWA concentrations of analytes during the deployment period is usually not practical with spot samples. The lower estimates by POCIS may be attributed to SPE's ability to extract both the freely dissolved and particle-bound fractions of the contaminants. On the other hand, POCIS targets only the freely dissolved portion. Several other extraneous environmental factors such as temperature changes, biofouling and water turbulence may not have been adequately accounted for in the laboratory calibration.
Estimates of aqueous diffusion coefficients (Dw) based on Hayduk and Laudie model, suggest a 30% increase in diffusivity and, hence, Rs for small molecule PPCPs when water (sample) temperature changes from 10 to 20°C and 75% increase when sample temperature change from 10 to 30°C (Macleod et al., 2007; Alvarez et al., 2005). In the current study, field temperatures were close to those used in the laboratory calibration, and serious fluctuations were not witnessed. As such, influence of temperature on field sampling rates may not have been a major factor.
Biofilm infestation enhances resistance to mass transfer of analytes in passive samplers (Vrana et al., 2005; Huckins et al., 2002). As the fouling layer is largely composed of organic material, its effect is therefore expected to be more significant for the less polar compounds. However, in the current case, the compounds studied had log Kow values less than 6, a figure proposed by Huckins et al. (2002) as the lower limit at which fouling by microorganisms is expected to significantly affect sampling rates of compounds. Moreover, PES membrane used in POCIS has been shown to considerably resist biofouling (Morin et al., 2012). Triclosan, being the most hydrophobic compound among the target compounds, was anticipated to suffer most from the effects of biofouling. This appeared not to have been the case. Although lower TWA analyte levels were obtained from POCIS in comparison with spot samples, there was no evidence linking increased hydrophobicity of the analyte with lower Rs. It was therefore concluded that the influence of biofouling on the results in this study may not have been severe. All in all, a greater understanding of the influence of membrane fouling on the uptake and, hence, Rs, of polar compounds by POCIS is still required.
The flow velocity prevailing at the sample site is known to significantly affect Rs (Booij et al., 2007). Actual field conditions encountered during deployment are usually not identical to laboratory conditions. Sampling rate is overestimated if the laboratory calibration value is too low (Macleod et al., 2007) and is underestimated if it is too high. Using the laboratory-derived sampling rates, TWA concentrations from POCIS were on average 40 to 65% lower than those obtained from grab samples. Among other factors, it is possible that during sampler deployment the hydrodynamic conditions in the field may not have been exactly similar to those used in the laboratory during calibration, and would, hence, have affected sampling rates. The ideal approach to overcoming this difficulty involves incorporation of performance reference compounds (PRCs), as has been the case in other passive samplers such as SPMDs (Vrana et al., 2005; Huckins et al., 2002). Unfortunately, suitable pharmaceutical PRCs that will significantly desorb from the POCIS are yet to be found (Morin et al., 2012).
Removal efficiency of the PPCPs in the WWTPs
The overall removal efficiency of the contaminants in the WWTPs was determined by considering the mean concentrations of each analyte in the raw influent and treated effluent. Average removal rates of the compounds ranged from 68 to 86% at Goudkoppies WWTP and 61 to 82% at Northern WWTP (Fig. 6). These findings were comparable to those reported elsewhere. In a study of similar compounds, Sagrista (2012) reported average removal efficiencies of 79%, 90% and 77% for naproxen, ibuprofen and triclosan, respectively. Lindqvist et al. (2005) and Zorita et al. (2009) also reported values that ranged between 81% and 94% for the two acidic pharmaceuticals.

CONCLUSIONS
This work assessed the viability of POCIS as a sampling technique for determining naproxen, ibuprofen, and triclosan in wastewater. Laboratory calibration of the samplers using a mixture of the three PPCPs was carried out under controlled conditions. Rs values that ranged between 0.087 and 0.383 ℓ.d-1, and 0.125 and 0.936 ℓ.d-1 in quiescent and stirred conditions, respectively, were obtained. Using these values, field performance of the POCIS in wastewater effluent yielded estimated concentrations of the compounds that were in the range of 10.7 to 13.5 µg.ℓ-1 in Goudkoppies WWTP and 20.4 to 24.6 µg.ℓ-1 in Northern WWTP. Influent PPCP concentrations in both treatment plants did not exceed 127.7 µg.ℓ-1. Comparison of POCIS-estimated concentrations with grab samples showed that levels from the latter were up to 3 times higher than in the former. This was attributed to the exhaustive nature of the SPE technique. However, these results showed that POCIS can provide a good assessment of the TWA concentrations of PPCPs in wastewater, and, especially, the freely dissolved fraction.
ACKNOWLEDGEMENTS
The authors are grateful to the management of Johannesburg Water for facilitating access to the sampling sites. The national Research Foundation (NRF) and Water Research Commission (WRC) are appreciated for their financial support.
REFERENCES
ALVAREZ DA, HUCKINS JN, PETTY JD, JONES-LEPP T, STUER- LAURIDSEN F, GETTING JP, GODDARD JP and GRAVELL A (2007) Passive sampling techniques in environmental monitoring. In: Greenwood R, Mills G and Vrana B (eds.) Comprehensive Analytical Chemistry 48. Elsevier, Amsterdam. 171-195. [ Links ]
ALVAREZ DA, STACKELBERG PE, PETTY JD, HUCKINS JN, FURLONG ET, ZAUGG SD and MEYER MT (2005) Comparison of a novel passive sampler to standard water-column sampling for organic contaminants associated with wastewater effluents entering a New Jersey stream. Chemosphere 61 610-622. [ Links ]
ALVAREZ DA, PETTY JD, HUCKINS JN, JONES-LEPP TL, GETTING DT, GODDARD JP and MANAHAN SE (2004) Development of a passive in situ, integrative sampler for hydro-philic organic contaminants in aquatic environments. Environ. Toxicol. Chem. 23 1640-1648. [ Links ]
BOOIJ K, VRANA B and HUCKINS JN (2007) Theory, modelling and calibration of passive samplers used in water monitoring. In: Greenwood R, Mills G and Vrana B (eds.) Comprehensive Analytical Chemistry 48. Elsevier, Amsterdam. 141-169. [ Links ]
BRAGA O, SMYTHE GA, SCHAFFER AI and FEITZ AJ (2005) Fate of steroid estrogens in Australian inland and coastal wastewater treatment plants. Environ. Sci. Technol. 39 3351-3358. [ Links ]
HUCKINS JN, PETTY JD, LEBO JA, ALMEIDA FV, BOOIJ K, ALVAREZ DA, CRANOR WL, CLARK RC and MOGENSEN BB (2002) Development of the permeability/performance reference compounds approach for in situ calibration of semipermeable membrane devices. Environ. Sci. Technol. 36 85-91. [ Links ]
JOHANNESBURG WATER (2012) Corporate profile, Johannesburg Water (Pty) Limited. URL: http://www.johannesburgwater.co.za (Accessed 11 April 2013). [ Links ]
JONES-LEPP TL, ALVAREZ DA and PETTY JD (2004) Polar organic chemical integrated sampling and liquid chromatography-electro-spray/ion-trap mass spectrometry for assessing selected prescription and illicit drugs in treated sewage effluent. Arch. Environ. Contam. Toxicol. 47 427-455. [ Links ]
KOT A, ZABIEGALA B and NAMIESNIK J (2000) Passive sampling for long-term monitoring of organic pollutants in water. Trends Anal. Chem. 19 (7) 446-459. [ Links ]
LI H, HELM PA and METCALFE DC (2010) Sampling in the Great Lakes for pharmaceuticals, personal care products, and endocrine-disrupting substances using the passive polar organic chemical integrative sampler. Environ. Toxicol. Chem. 29(4) 751-762. [ Links ]
LINDQVIST N, TUHKANEN T and KRONBERG L (2005) Occurrence of acidic pharmaceuticals in raw and treated sewages and in receiving waters. Water Res. 39 2219-2228. [ Links ]
MACLEOD SL, McCLURE EL and WONG CS (2007) Laboratory calibration and field deployment of the polar organic chemical integrative sampler for pharmaceuticals and personal care products in wastewater and surface water. Environ. Toxicol. Chem. 26 2517-2529. [ Links ]
MAZZELLA N, DUBERNET JF and DELMAS F (2007) Determination of kinetic and equilibrium regimes in the operation of polar organic chemical integrated samplers - application to the passive sampling of the polar herbicides in the aquatic environments. J. Chromatogr. A 1154 42-51. [ Links ]
MIAO X-S, BISHAY F, CHEN M and METCALFE CD (2004) Occurrence of antimicrobials in the final effluents of wastewater treatment plants in Canada. Environ. Sci. Technol. 38 3533-3541. [ Links ]
MORIN N, MIEGE C, COQUERY M and RANDON J (2012) Chemical calibration, performance, validation and applications of polar organic integrative sampler (POCIS) in aquatic environments. Trends Anal. Chem. 36 144-175. [ Links ]
SABALIUNAS D and SODERGREN A (1997) Use of semipermeable membrane devices to monitor pollutants in water and assess their effects: A laboratory test and field verification. Environ. Pollut. 96 (2) 195-205. [ Links ]
SAGRISTA E (2012) Hollow fibre liquid-phase microextraction in the determination of pharmaceuticals and personal care products. PhD thesis, Universitat de Girona, Spain. [ Links ]
TOGOLA A and BUDZINSKI H (2007) Development of polar organic integrative samplers for analysis of pharmaceuticals in aquatic systems. Anal. Chem. 79 6734-6741. [ Links ]
VRANA B, MILLS GA, ALLAN IJ, DOMINIAK E, SVENSSON K, MORRISON G and GREENWOOD R (2005) Passive sampling techniques for monitoring pollutants in water. Trends Anal. Chem. 24 845-868. [ Links ]
VRANA B and SCHUURMANN G (2002) Calibrating the uptake kinetics of semipermeable membrane devices in water: impact of hydrodynamics. Environ. Sci. Technol. 36 290-296. [ Links ]
ZORITA S, MÅRTENSSON L and MATHIASSON L (2009) Occurrence and removal of pharmaceuticals in a municipal sewage treatment system in the south of Sweden. Sci. Total Environ. 407 2760-2770. [ Links ]
 Correspondence:
Correspondence:
Luke Chimuka
+27(11) 717 6703, +27(72) 497 8041; fax: +27(11) 717 6749
e-mail: Luke.Chimuka@wits.ac.za
Received 24 June 2013
Accepted in revised form 2 May 2014


{kind=link}
{kind=link}