










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkWater SA
On-line version ISSN 1816-7950
Print version ISSN 0378-4738
Water SA vol.39 n.5 Pretoria Jan. 2013
Detection and confirmation of toxigenic Vibrio cholerae 01 in environmental and clinical samples by a direct cell multiplex PCR
JP Yadava; M Jain; AK Goel*
Biotechnology Division, Defence Research & Development Establishment, Jhansi Road, Gwalior-474 002, India
ABSTRACT
Epidemic cholera caused by toxigenic Vibrio cholerae O1 is a major health problem in several developing countries. Traditional methods for identifying V. cholerae involve cultural, biochemical and immunological assays which are cumbersome and often take several days to complete. In the present study, a direct cell multiplex PCR was developed targeting the ompW, ctxB and rfbO1 genes for confirmation of V. cholerae, its toxigenicity and serogroup Ol, respectively from clinical and environmental samples. The detection sensitivity of the multiplex PCR was 1.9 x 103V. cholerae per PCR reaction. A total of 31 environmental samples and 45 clinical V. cholerae isolates from different outbreaks were examined by the PCR. The assay was simple and specific, as there was no requirement for DNA extraction and no amplification was observed with other homologous bacteria used. The assay can be very useful for rapid surveillance of toxigenic V. cholerae O1 in environmental water samples, as well as for confirmation of clinical isolates.
Keywords: cholera, Vibrio cholerae, PCR, environmental samples
INTRODUCTION
Cholera is an epidemic diarrheal disease that continues to devastate many countries where primary health and sanitation is still a challenge. Several cholera episodes are reported from developing countries due to contamination of water supplies. In 2010, a total of 48 countries reported 317 534 cases of cholera to the WHO, of which 36% were reported from Africa and 56.6% from the Americas, where a large outbreak was reported in Haiti (WHO, 2011). A total of 5 155 cases including 9 deaths were reported from India (WHO, 2011). Globally, the true number of cholera cases may be much higher because many cholera cases remain unreported. Cholera is caused by Vibrio cholerae, which is primarily an inhabitant of aquatic environments (Faruque et al., 1998, Islam et al., 1994). Therefore, water plays a significant role in the transmission and epidemiology of cholera. Monitoring the presence of V. cholerae in drinking water sources prior to disease outbreak is important to enable effective resource management and public health protection (Choopun et al., 2002).
V. cholerae is classified into more than 200 serogroups, but only V. cholerae O1 and O139 that produce cholera toxin (CT) have been associated with epidemics and pandemics (Rivera et al., 2001). The other non-O1, non-O139 serogroups are usually associated with some cases of mild gastroenteritis (Kaper et al., 1995). The ability to produce CT has been used to distinguish between choleragenic and non-choleragenic strains. CT non-producer O1 and O139 (non-choleragenic) strains do not cause epidemic cholera (Levine et al., 1983); and CT-producing non-O1/non-O139 strains, although pathogenic, lack epidemic potential. Hence, detection and monitoring of toxigenic V. cholerae O1 or O139 is important during surveillance.
Various conventional cultural-, biochemical- and immu-nological-based assays are used for detection of V. cholerae. However, many of these methods are time-consuming and laborious. Moreover, biochemical identification systems may not always be accurate as several Vibrio species display similar biochemical characteristics (Nishibuchi, 2006; O'Hara et al., 2003). Several investigators have employed a direct immunofluorescence technique using polyclonal or monoclonal antibodies for detection of various bacteria and spores (Goel et al., 2005a; Huq et al., 1990). In contrast to culturing and immunological techniques, molecular methods have several advantages: they are rapid, sensitive, highly selective and do not require extensive hands-on time. In this study, we have developed a simple, sensitive and specific multiplex PCR that confirms the presence of V. cholerae in environmental and clinical samples, and exhibits its epidemic potential.
MATERIALS AND METHODS
Bacterial strains
The reference strain of Vibrio cholerae 20 O1 Ogawa was procured from the National Institute of Cholera and Enteric Disease (NICED), Kolkata, India, and was maintained on Brain Heart Infusion (BHI) agar (Difco, USA) at 22 °C. The other bacterial strains used in this study were V. cholerae O1 El Tor N16961 (ATCC 39315), V. cholerae O1 El Tor (ATCC 14033), V. cholerae O1 Classical 569B(ATCC 25870), V. cholerae O1 Classical (ATCC 11623), V. cholerae O139 (ATCC 51394), V. fischeri (MTCC 1738), V. parahaemolyticus (MTCC 451), Salmonella typhimurium (MTCC 98), S. paratyphi A (MTCC 735), S. virchow (MTCC 1163), S. infantis (MTCC 1167), S. bovismorficans (MTCC 1162), Shigella sonnei (MTCC 2957), Shigellaflexneri (MTCC 1457), Shigella dysenteriae (NICED isolate), Escherichia coli (ATCC 11775) and Bacillus cereus(ATCC 10876).
Determination of sensitivity of direct cell PCR
Vibrio cholerae 20 O1 was grown in Brain Heart Infusion (BHI) broth for 16-18 h at 37°C and 10-fold serial dilutions were made in PBS, pH 7.4. Bacterial enumeration to determine CFU/µℓ was done by plating 100 µℓ of each dilution on BHI agar plates, followed by incubation at 37°C for 18 h. A 1-µℓ culture from each dilution was washed twice with de-ionised water by centrifugation at 10 000 x g for 10 min and finally re-suspended in 1 µℓ de-ionised water. Five microlitres (5 µℓ) of each dilution was used directly as template in PCR.
Processing of clinical samples
Clinical samples were processed for isolation of V. cholerae as described previously (Pourshafie et al., 2007). V. cholerae isolates from different outbreaks were grown on BHI agar plate for 16-18 h at 37°C and a loopful of bacterial culture was suspended in 500 µℓ de-ionised water. After mixing well, 5 µℓ of the solution was used directly in PCR. A total of 45 isolates from 3 different cholera outbreaks (15 isolates from each outbreak) in India were used in this study.
Processing of environmental water samples
A total of 31 water samples were collected from surface water (10), groundwater (10) and sewage water (11) from in and around Gwalior, Madhya Pradesh, India (26°13' N; 78°13' E). A total of 100 µℓ of water sample was filtered through a polycarbonate membrane filter of 0.45 ìm pore size using vacuum pressure of 103-137 kPa. Sewage samples (100 µℓ) were mixed with an equal volume of distilled water, mixed well and allowed to settle. Slurry from the upper surface was processed for filtration. After this, the membranes were incubated in 5 µℓ of alkaline peptone water (APW, 1% peptone (w/v), 1% NaCl (w/v), pH 8.6) for 6 h at 37 ± 1°C under shaking conditions (100 r/min). One millilitre (1 µℓ) of enriched culture broth, taken from the top layer of APW was centrifuged at 10 000 x g for 10 min to collect the bacterial cells. The pellet was washed twice with de-ionised water and finally suspended in 20 µℓ of de-ionised water; 5 µℓ of the suspension was used for PCR. Negative, un-inoculated water samples were included as controls in each of the experiments. Simultaneously, all of the APW enriched samples were also grown on TCBS agar plates for confirmation of V. cholerae. Two loopfuls of the APW culture broth were streaked on to thiosulphate bile salt sucrose (TCBS) agar plates (Difco, USA) and incubated overnight at 37 ± 1°C. The yellow, flat colonies of 1-3 mm diameter were considered as V. cholerae (Tamrakar et al., 2006)
PCR
Three sets of primers for genes encoding the outer-membrane protein OmpW (ompW), cholera toxin (ctxB) and O1 somatic antigen (rfbO1) were used in this study. The sequences of primers (5'-3') used were:
ompW (For: CACCAAGAAGGTGACTTTATTGTG;
Rev: GAACTTATAACCACCCGCG);
ctxB (For: GCACATGGAACACCTCAA;
Rev: GGCAAAACGGTTGCTTCT) and
rfbO1 (For: TCTATGTGCTGCGATTGGTG;
Rev: CCCCGAAAACCTAATGTGAG).
The primers targeting the genes encoding ompW, ctxB and rfbO1 amplify fragments of 588 bp, 386 bp and 638 bp, respectively. Primers were selected from previous studies (Nandi et al., 2000) or were designed on the basis of GenBank gene sequences of V. cholerae using Oligo Explorer version 1.2 (Gene Link). In the PCR assay, bacterial cells suspended in de-ionised water were used as template for amplification. To determine the specificity of the PCR assay, a single colony of other bacteria strains mentioned above were suspended in 500 µℓ de-ionised water and used in the PCR. Vibrio cholerae cells were subjected to triplex PCR assay by simultaneous addition of the primer pairs for ompW, ctxB and rfbO1 in the same reaction mixture. PCR amplification of the target DNA was carried out in a 200-µℓ PCR tube with a reaction mixture of 25 µℓ. Each of the reaction mixtures contained 1 x master mix (Fermentas, USA), 10 pmol each of the ompW and rfbO1 primers, 20 pmol of the ctxB primers, 5 µℓ of template (direct cells), and distilled water. The master mix was supplied as a 2 x concentrated solution containing Taq DNA polymerase, reaction buffer, MgCl2 and dNTPs. Amplifications were carried out in a thermal cycler (Corbett Life Sciences, Australia) programmed for 30 three-step cycles starting with denaturation of the template DNA at 95°C for 45 s; annealing at 57°C for 45 s and extension of the primers at 72°C for 1 min. Before initiation of the first cycle, the reaction mixture was heated at 94°C for 10 min to allow complete denaturation of the template. After the last cycle, the reaction mixture was subjected to 72°C for 10 min to ensure final extension. In a control PCR reaction, deionised water was added to the reaction mixture instead of bacterial cells. PCR products thus obtained were electro-phoresed on a 1.8% (w/v) agarose gel containing ethidium bromide (0.5 µg∙mℓ-1) to resolve the amplified products. A 100 bp DNA ladder (Fermentas, USA) was loaded onto the agarose gel as a molecular size standard.
RESULTS AND DISCUSSION
Multiplex PCR enables simultaneous amplification of many targets of interest in one reaction by using more than one pair of primers. The isolates were screened for the presence of 3 different genes for identification, toxigenicity and serogroup of V. cholerae. PCR sensitivity was determined by using 10-fold serially diluted V. cholerae cells for amplification of the ompW, ctxAB and rfbO1 genes. Amplified fragments of 588, 386 and 638 bp could be visualised on 1.8% agarose with ethidium bromide in a reaction mixture that contained 1.9 x 103 CFU/ reaction i.e 3.8 x 105 CFU/µℓ (Fig. 1). The results showed that direct bacterial cells of a toxigenic V. cholerae culture could readily serve as template for PCR and there is no need for DNA extraction. Cell lysis during the initial denaturation at 94°C for 10 min during PCR is sufficient to provide a suitable template.

In the multiplex PCR, all the reference isolates of V. cholerae, except V. cholerae O1 El Tor (ATCC 14033) and V. cholerae O139 (ATCC 51394), showed all three genes (Table 1, Fig. 2). V. cholerae O1 El Tor ATCC 14033 does not possess the ctxB gene and hence was PCR positive only for the ompW and rfbO1 genes. Likewise, V. cholerae O139 (ATCC 51394) does not possess the rfbO1 gene and hence was PCR positive only for ompW and ctxB genes (Fig. 2). The multiplex PCR was specific as no amplification was observed with other bacteria used in the study (Table 1).
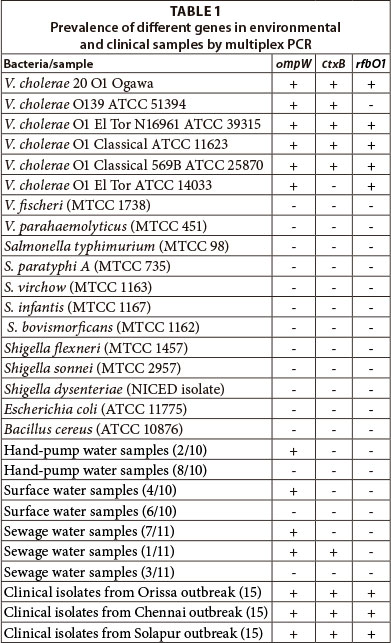

The ompW gene is species-specific for V. cholerae and its presence confirmed that the isolates were V. cholerae. Nucleotide sequence data has shown that the ompW sequence is highly conserved among V. cholerae strains belonging to different biotypes or serotypes (Nandi et al., 2000). The presence of the ctxB and rfbO1 genes confirmed the toxigenicity and O1 serogroup of the isolates, respectively. The cholera toxin is considered the most important epidemic marker among various toxins produced by V. cholerae (Kaper et al., 1995). The presence of the somatic O-antigen biosynthesis gene rfbOl provided molecular evidence for O1 serogroups, which has epidemic and pandemic potential.
A total of 31 water samples collected from different locations and 45 V. cholerae isolates from 3 different cholera outbreaks in India were subjected to multiplex PCR. Out of 31 environmental samples, only 14 samples tested positive for bacteria containing ompW gene (Table 1). One sewage water sample tested positive for ompW and ctxB gene, indicating the presence of toxigenic V. cholerae (Table 1). However, the rfbOl gene was not present in any of the samples, indicating the presence of non-O1 V. cholerae in the water samples. Prior to PCR, environmental water samples were filtered and enriched in APW, which specifically promotes the growth of Vibrio spp. due to its alkaline pH. The enriched samples were centrifuged and washed with deionised water and directly used in PCR. Enrichment of V. cholerae in APW for 6 h was sufficient to multiply the bacteria to within the limits of sensitivity of PCR. In earlier studies, it was found that about a single CFU/µℓ of V. cholerae could be detected in water after 6 h enrichment in APW (Goel et al., 2005b). All 14 samples tested positive on TCBS plates and using other biochemical tests, which indicated that environmental strains contained non-O1 V. cholerae. The 45 clinical V. cholerae isolates from different cholera outbreaks all harboured the three genes i.e. ompW, ctxB and rfbO1 (Table 1). The study thus indicated that the clinical V. cholerae isolates possess the cholera toxin gene and generally belong to the O1 serogroup. In contrast, most of the environmental samples contained non-toxigenic, non-O1 V. cholerae.
Traditional methods for cholera diagnosis are time consuming and further do not confirm the toxigenicity of strains. PCR has become an important and powerful tool for the detection of pathogenic micro-organisms. However, DNA isolation is a cumbersome process in PCR. Hence, the present PCR assay obviates the extraction of DNA and bacteria are rather used as PCR template. Therefore, the multiplex PCR assay described herein is a quick, sensitive and effective method for monitoring of V. cholerae in the environment as well as for confirmation of its toxigenicity and epidemic potential.
CONCLUSION
Toxigenic Vibrio cholerae O1 is the major causative agent of cholera. In this study, a simple, sensitive and specific multiplex PCR has been designed that confirms V. cholerae, its toxigenicity and serogroup O1. The assay obviates the need for extraction of DNA and the enriched environmental samples or clinical isolates can be used directly as PCR templates. Thus, the present assay can be a useful tool for monitoring the presence V. cholerae in environmental water samples.
ACKNOWLEDGEMENTS
Authors are thankful to Director, Defence Research & Development Establishment (DRDE), Gwalior for providing necessary facilities and funds for the work.
REFERENCES
CHOOPUN N, LOUIS V, HUQ A and COLWELL RR (2002) Simple procedure for rapid identification of Vibrio cholerae from the aquatic environment. Appl. Environ. Microbiol. 68 (2) 995-998. [ Links ]
FARUQUE SM, ALBERT MJ and MEKALANOS JJ (1998) Epidemiology, genetics, and ecology of toxigenic Vibrio cholerae. Microbiol. Mol. Biol. Rev. 62 (4) 1301-1314. [ Links ]
GOEL AK, TAMRAKAR AK, KAMBOJ DV and SINGH L (2005a) Direct immunofluorescence assay for rapid environmental detection of Vibrio cholerae O1. Folia Microbiol. (Praha) 50 (5) 448-452. [ Links ]
GOEL AK, TAMRAKAR AK, NEMA V, KAMBOJ DV and SINGH L (2005b) Detection of viable toxigenic Vibrio cholerae from environmental water sources by direct cell duplex PCR assay. World J. Microbiol. Biotechnol. 21 (6-7) 973-976. [ Links ]
HUQ A, COLWELL RR, RAHMAN R, ALI A, CHOWDHURY MA, PARVEEN S, SACK DA and RUSSEK-COHEN E (1990) Detection of Vibrio cholerae O1 in the aquatic environment by fluorescent-monoclonal antibody and culture methods. Appl. Environ. Microbiol. 56 (8) 2370-2373. [ Links ]
ISLAM MS, HASAN MK, MIAH MA, YUNUS M, ZAMAN K and ALBERT MJ (1994) Isolation of Vibrio cholerae O139 synonym Bengal from the aquatic environment in Bangladesh: implications for disease transmission. Appl. Environ. Microbiol. 60 (5) 1684-1686. [ Links ]
KAPER JB, MORRIS JG, JR. and LEVINE MM (1995) Cholera. Clin. Microbiol. Rev. 8 (1) 48-86. [ Links ]
LEVINE MM, KAPER JB, BLACK RE and CLEMENTS ML (1983) New knowledge on pathogenesis of bacterial enteric infections as applied to vaccine development. Microbiol. Rev. 47 (4) 510-550. [ Links ]
NANDI B, NANDY RK, MUKHOPADHYAY S, NAIR GB, SHIMADA T and GHOSE AC (2000) Rapid method for species-specific identification of Vibrio cholerae using primers targeted to the gene of outer membrane protein OmpW. J. Clin. Microbiol. 38 (11) 4145-4151. [ Links ]
NISHIBUCHI M (2006) Molecular Identification. In: Thompson FL, B Austin and J Swingss (eds.) The Biology of the Vibrios. ASM Press, Washington, DC. 44-66. [ Links ]
O'HARA CM, SOWERS EG, BOPP CA, DUDA SB and STROCKBINE NA (2003) Accuracy of six commercially available systems for identification of members of the family vibrionaceae. J. Clin. Microbiol. 41 (12) 5654-5659. [ Links ]
POURSHAFIE MR, BAKHSHI B, RANJBAR R, SEDAGHAT M, SADEGHIFARD N, ZAEMI YAZDI J, PARZADEH M and RAESI J (2007) Dissemination of a single Vibrio cholerae clone in cholera outbreaks during 2005 in Iran. J. Med. Microbiol. 56 (Pt 12) 1615-1619. [ Links ]
RIVERA IN, CHUN J, HUQ A, SACK RB and COLWELL RR (2001) Genotypes associated with virulence in environmental isolates of Vibrio cholerae. Appl. Environ. Microbiol. 67 (6) 2421-2429. [ Links ]
TAMRAKAR AK, GOEL AK, KAMBOJ DV and SINGH L (2006) Surveillance methodology for Vibrio cholerae in environmental samples. Int. J. Environ. Health Res. 16 (4) 305-312. [ Links ]
WHO (2011) Cholera, 2010. Wkly. Epidemiol. Rec. 86 (31) 325-339. [ Links ]
Received 18 June 2012
Accepted in revised form 20 August 2013
* To whom all correspondence should be addressed. +91-751-2233742; fax: 91-751-2341148; e-mail: akgoel73@yahoo.co.uk

