










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkWater SA
versión On-line ISSN 1816-7950
versión impresa ISSN 0378-4738
Water SA vol.39 no.3 Pretoria 2013
Development and implementation of a rapid real-time polymerase chain reaction assay for the detection of toxigenic Vibrio cholerae in water
Neil Leat*; Monique Grundlingh
Microbiology Laboratory, Analytical Services, Rand Water, PO Box 3526, Vereeniging, 1930, South Africa
ABSTRACT
Assays which use real-time polymerase chain reaction (PCR) technology can be developed for the rapid identification of genetic sequences carried by waterborne pathogens. Rand Water has established facilities within which a selection of PCR assays will be developed. This paper reports on the optimisation and validation of the first assay to be implemented. This assay facilitates the detection of the ctxA gene of toxigenic Vibrio cholerae (V. cholerae) strains. The assay also includes an internal process control in the form of an Escherichia coli (E. coli) strain carrying a single genomic copy of the gfp gene from Aequorea victoria. Establishment of the assay required the selection of suitable PCR primers and probes for both the ctxA and gfp genes. This was followed by an optimisation phase where ideal PCR cycling conditions and primer/probe concentrations were established. A validation phase established the performance parameters of the assay. Parameters assessed included: limit of detection, sensitivity, specificity, reproducibility and robustness. The validation was conducted using potable water, surface water and sewage effluent matrices. The process has resulted in the establishment of a robust assay for the detection of toxigenic V. cholerae strains within 24 hours after samples are received.
Keywords: real-time PCR, Vibrio cholerae O1, ctxA gene, validation
INTRODUCTION
Close to 200 serotypes of V. cholerae have been identified; however, significant cholera epidemics have been associated almost exclusively with the toxigenic Ol and O139 serotypes (Kaper et al., 1995). Ingestion of toxigenic V. cholerae results in severe watery diarrhoea and may be fatal in the absence of suitable medical intervention. While it would be unlikely for V. cholerae to bypass well-managed potable water treatment systems, rigorous monitoring provides additional confidence that safe water is being produced.
To date Rand Water has used a conventional culture-based approach for the identification of V. cholerae in water samples. This approach involves enrichment of Vibrio species in samples using alkaline peptone water (APW), with incubation at 36°C for 16 to 18 h. This is followed by isolation of presumptive Vibrio cholerae strains on thiosulfate citrate bile salts sucrose agar (TCBS). Presumptive isolates are confirmed using biochemical tests followed by a serological assessment to identify the V. cholerae Ol antigen. While this process has been effectively implemented, it takes a number of days to complete.
Real-time polymerase chain reaction (PCR) technology provides a more efficient alternative to the classical culture-based approach. The key advantage is that virulence genes can be directly and rapidly detected in APW enrichments. To date, a plethora of real-time PCR assays for the detection of V. cholerae have been published. Three examples are the assays published by Huang et al. (2009), Koskela et al. (2009) and Mehrabadi et al. (2012). The assay published by Huang et al. (2009) is a quadruplex real-time PCR assay targeting 4 genes (ctxA, hlyA, O1-rfb and O139- rfb). The assay published by Koskela et al. (2009) targets 2 virulence genes (i.e. toxR activator and ctxA). The assay published by Mehrabadi et al. (2012) targets the ctxA, tcpA and ompW genes.
While the elaborate multiplex assays typically published in the literature are technologically impressive, almost none of these include a single process control which is carried through both nucleic acid extraction and PCR amplification steps. In addition, most target several virulence genes which may introduce unnecessary complexity when the principal concern is to detect toxigenic V. cholerae in the simplest way possible.
The requirement within Rand Water was to establish a simple robust assay that would target one toxigenic V. cholerae virulence gene as well as one process control gene, carried through both nucleic acid extraction and PCR amplification steps. By selecting the simplest assay possible the subsequent validation and implementation of the assay would be more readily achieved. For the purposes of this study the ctxA gene was selected as the primary target. The ctxA gene encodes the cholera toxin and is one of the most important factors responsible for severe diarrhoea Kaper et al. (1995).
For the present study a process control was developed based on the system described by Murphy et al. (2007). The process control utilises an Escherichia coli BW 31004 strain which has been engineered to carry a single copy of the gfp gene on its genome (Zhou et al., 2004). The gfp gene encodes the green fluorescent protein of the jellyfish Aequorea victoria. The gfp gene was considered unlikely to be present in environmental samples analysed by Rand Water.
The implementation of the current assay required the selection of suitable PCR primers and probes for both the primary target gene (ctxA) and the process control gene (gfp). This was followed by an optimisation phase where ideal PCR cycling conditions and primer/probe concentrations were determined. A validation phase established the performance parameters of the assay. The purpose of this paper is to describe the optimised assay conditions and present findings from selected validation experiments. It is important to note that the assay was designed to be used on alkaline peptone water enrichments to yield a presence/absence result for the ctxA gene.
MATERIALS AND METHODS
Bacterial strains and culture media
Alkaline peptone water (APW) (Oxoid CM1028) was used to enrich Vibrio species in water samples. Enrichments were incubated for 16 to 18 h at 36°C. The process control strain (E. coli BW31004) was spiked into enriched samples immediately before nucleic acid extraction. The volume of process control added was calculated to achieve a final concentration of 104 colony forming units/mℓ (cfu/mℓ). Subsequent detection of the gfp gene was regarded as an indication that substantial PCR inhibition had not taken place.
Some validation experiments required that the samples be spiked with V. cholerae O1 before APW enrichment. This was achieved using dilutions of a pure V. cholerae O1 NCTC 8021 culture grown in nutrient broth (Oxoid CM0067). In such cases the number of organisms spiked into each sample was estimated by plating dilutions of the pure V. cholerae O1 culture on nutrient agar. The number of colonies on each plate was counted after overnight incubation at 37°C.
Some validation experiments required that the samples be spiked with V. cholerae O1 after APW enrichment. This was achieved using a frozen V. cholerae O1 NCTC 8021 culture. The concentration of V. cholerae O1 was estimated using nutrient agar plate counts before it was frozen. When required, aliquots of the stock were thawed and used to spike enriched samples.
Nucleic acid extraction from APW enrichments
In order to detect the presence of the ctxA and gfp genes, DNA was extracted from 1 mℓ aliquots of APW enrichments. This was achieved by pelleting bacterial cells using centrifuga-tion (Eppendorf 5424 microcentrifuge, 14 000 r/min for 10 min) followed by aspiration of the supernatant. Cell pellets were resuspended in 100 µℓ volumes of Applied Biosystems PrepMan Ultra Sample Preparation Reagent and heated at 100°C for 10 min. For each extract, 20 µℓ was diluted in 80 µℓ nuclease free water prior to being added to PCR reactions. This final dilution step was introduced after consistently encountering inhibition with undiluted extracts during initial optimisation of the assay. After the introduction of the dilution step, consistent amplification of the gfp process control was observed for all samples, suggesting sufficient dilution of associated PCR inhibitors.
Polymerase chain reaction conditions
PCR reactions were conducted in 25 µℓ£ volumes in 0.1 mℓ Qiagen PCR tubes. Each PCR reaction contained Applied Biosystems TaqMan* Environmental Master Mix (12.5 µℓ), 10x primer and probe stock (2.5 µℓ); Applied Biosystems AmpErase* Uracil N-glycosylase (0.25 µℓ), Qiagen nuclease free water (4.75 µℓ) and the relevant nucleic extract being tested (5 µℓ). The primers and probes used in the study were synthe-sised by Sigma-Aldrich and supplied at a concentration of 200 µM in liquid form. Details of the primers and probes used in the optimised assay are presented in Table 1.
PCR reactions were run on a Rotor-Gene Q real-time PCR instrument with the following thermocycling profile: 50°C for 2 min; 95°C for 10 min and 50 cycles of 95°C for 20 s and 63°C for 30 s. Fluorescence was recorded in the green and yellow channels corresponding to emissions from the Fam and Vic dyes, respectively. Fluorescence in the green channel represented an accumulation of the ctxA PCR product while fluorescence in the yellow channel represented an accumulation of the gfp PCR product. The build-up of fluorescence for each sample was recorded as a cycle threshold (Ct) value. The cycle threshold value is the number of PCR cycles required before the fluorescence signal in a reaction exceeds a previously established fluorescence threshold. During the implementation phase of the assay the ctxA gene was considered to be present in the nucleic acid extract from an APW enrichment, if the corresponding Ct value was between 1 and 45 cycles. Ct values above 40 cycles were seldom encountered.
RESULTS AND DISCUSSION
The real-time PCR assay being considered was designed to detect the key virulence gene ctxA, found in pathogenic strains of V. cholerae, and the gfp gene carried by the process control strain E. coli BW31004. The assay was designed to be used on alkaline peptone water (APW) enrichments of potable water, surface water and sewage effluent samples. The optimised assay conditions are summarised above. What follows are selected findings from validation experiments conducted using the optimised assay.
Detection limit for viable V. cholerae O1 cells spiked before APW enrichment
Determining the limit of detection is often a necessary step when assessing the utility of an assay. However, APW enrichment makes the assessment challenging. This is for two reasons: firstly, environmental isolates of V. cholerae may be viable but not readily culturable, and, secondly, competing Vibrio species in a sample may limit the growth of the toxigenic V. cholerae present. It is almost impossible to predict what proportion of viable V. cholerae in a sample can be cultured or how many competing species will be present. Therefore, the number of toxigenic V. cholerae enriched may not always correlate with the number in the original sample.
Despite the difficulties, an attempt was made to demonstrate that the assay was able to enrich and detect low numbers of V. cholerae O1 in potable water, surface water and sewage effluent samples. In order to achieve this, a composite sample was prepared for each matrix from 5 individual samples. Each composite was then divided into five 100 mℓ sub-samples. Before APW enrichment each sub-sample was spiked with a 100 µℓ aliquot from a diluted V. cholerae O1 culture. Estimates of the numbers of V. cholerae spiked into each sub-sample were made using nutrient agar plate counts. Unspiked samples were processed in parallel as negative controls.
The estimated number of V. cholerae O1 cells spiked into each matrix sub-sample was as follows: i) 3 x 105; ii) 3 x 104; iii) 3 000; iv) 300 and v) 30 cfu. In every instance where V. cholerae O1 was spiked into a sample clear amplification of the ctxA gene was observed. This is illustrated by the amplification results for the spiked sewage effluent samples presented in Fig. 1. Equivalent results were observed for the potable water and surface water matrices. The experiment demonstrates that when 30 viable V. cholerae O1 cells are present per 100 mℓ of sample they will be effectively enriched using APW and the ctxA gene will readily be detected.
It is important to note that while the assay has been demonstrated to detect at least 30 viable V. cholerae O1 cells per 100 mℓ, the actual limit of detection is likely to be lower. The reason this could not be investigated is because it is challenging to consistently prepare spiked samples with less than 30 viable cells per 100 mℓ. This is a practical constraint around the accurate preparation of spiked samples with very low concentrations of the target species.
Sensitivity and specificity
It is essential that assays distinguish between samples containing the target organism and those that do not. This property of a method is readily assessed using the following parameters:
diagnostic sensitivity, diagnostic specificity, false positive rate and false negative rate. These parameters are described by
Eqs. (1) to (4) below.
Diagnostic sensitivity: Proportion of positive targets correctly assigned by the method.

Diagnostic specificity - proportion of negative targets correctly assigned by the method:
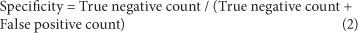
False positive rate - fraction of positive results that are wrongly assigned:

False negative rate - fraction of the negative results that are wrongly assigned:
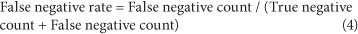
The experiment to investigate sensitivity and specificity utilised samples collected over a number of days. The samples were enriched using APW and then frozen. This approach meant that a broad selection of samples could be enriched and stored before being analysed using PCR.
A total of 29 potable water, 30 surface water and 30 sewage effluent samples were collected, enriched and frozen. On days when PCR analysis was conducted samples were thawed and split into two 1 mℓ aliquots. One aliquot was spiked with a previously quantified frozen stock of V. cholerae O1 to a final concentration of approximately 5 000 cfu/mℓ. This was done to simulate an effective APW enrichment of V. cholerae O1. The other served as an unspiked control. The split samples were analysed and the results were categorised per matrix as follows:
- True positives: Spiked samples where clear amplification of the ctxA gene is observed
- False positives: Unspiked samples where clear amplification of the ctxA gene is observed
- True negatives: Unspiked samples without evidence of ctxA amplification
- False negatives: Spiked samples without evidence of ctxA amplification
Irrespective of the matrix involved, every spiked sample generated clear evidence of ctxA amplification while no ctxA amplification was observed for unspiked samples. It follows that for the matrices tested the values determined for sensitivity and specificity equalled the ideal of 1. Similarly, the false positive and false negative rates equalled the ideal value of zero. The data indicate that, where the APW enrichment is effective and V. cholerae numbers reach at least 5 000 cfu/mℓ after enrichment, the real-time PCR assay under study will rapidly and effectively identify the presence of the ctxA gene in the sample.
Reproducibility
The reproducibility of the method is an indication of the degree of consistency between the results for the same sample tested under different conditions. Assessment of reproducibility often involves the repeated analysis of the same sample over an extended period of time by different analysts. This is often challenging to achieve using microbiological samples, which are typically unstable. However, since the method under study detected the ctxA gene rather than viable V. cholerae cells it was possible to prepare APW enrichments spiked with V. cholerae O1 and store them at -20 °C for analysis over several days. While the V. cholerae O1 cells spiked into each enrichment would most likely not be viable after storage, the ctxA genes would almost certainly be stable and readily detected. Analysis of the frozen samples would provide an indication of the reproducibility of the real-time PCR assay for the ctxA gene.
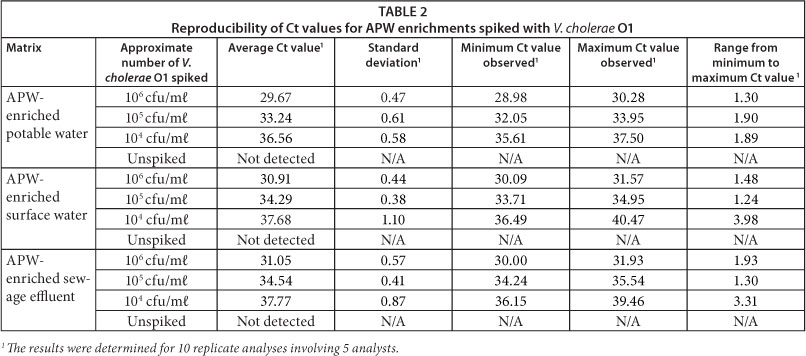
For the execution of the experiment, sewage effluent, potable water and surface water samples were enriched using APW with incubation at 36°C for 16 to 18 h. Each enrichment was divided into 3 volumes and spiked with a previously quantified frozen stock of V. cholerae O1 to achieve concentrations of 106, 105, 104 cfu/mℓ. The spiked samples were then stored as 1 mℓ aliquots at -20 °C for analysis. Unspiked aliquots from each enrichment were also stored to confirm that endogenous V. cholerae O1 cells were not present in the samples.
Every sample was analysed at least 10 times on different days. Five different analysts were involved in the execution of the experiment. Table 2 provides an indication of the reproducibility of cycle threshold (Ct) values observed for each matrix containing different amounts of V. cholerae O1. The last column of the table gives an indication of the difference between the minimum and maximum Ct values observed for the ten replicate analyses conducted for each sample. The relatively narrow margin observed suggests the assay can be reproduced with sufficient consistency for a presence/absence method.
Robustness
Among the robustness parameters investigated, an assessment was made of the impact of pipetting on PCR reactions. In order to do this the variation in delivery volumes for the relevant micropipettes was assessed. The variation was expressed as a standard deviation determined from 10 replicate measurements of the relevant delivery volume. Once this was determined PCR reaction mixtures were deliberately manipulated to prepare a concentrated PCR reaction mixture and a dilute PCR reaction mixture. The concentrated mixture corresponded to a PCR reaction in which the components were added with pipettes set to volumes 3 standard deviations above the normal volume. The dilute mixture corresponded to a PCR reaction in which the components were added with pipettes set to volumes corresponding to 3 standard deviations below the normal volume.
A composite matrix was prepared by mixing a sewage effluent, potable water and surface water sample. The sample was enriched using APW and incubation at 36°C for 16 to 18 h. The sample was then spiked with V. cholerae O1 to a final concentration of approximately 5 000 cfu/mℓ to simulate an effective enrichment. Nucleic acids were extracted from 10 replicate aliquots of the spiked enrichment and analysed using the concentrated and dilute PCR reaction mixtures described above.
The mean Ct value observed for the dilute PCR reaction mixture was 36.18 with a standard deviation of 0.19 cycles. The mean Ct value observed for the concentrated PCR reaction mixture was 37.22 with a standard deviation of 0.47 cycles. While there appears to be a difference between the Ct values observed for the dilute and concentrated PCR master mixtures the average difference is close to 1 cycle. Given that the method will be applied in a presence/absence format it seems unlikely that pipetting error during the preparation of PCR reactions will substantially influence the results obtained.
CONCLUSIONS
A real-time assay was designed for the detection of the ctxA and gfp genes. The ctxA gene represents the most significant virulence determinant from toxigenic V. cholerae strains, while the gfp gene is carried by the E. coli BW31004 process control strain. Optimised PCR conditions were presented as part of this report. In addition, results from selected validation experiments were presented. The results of these validation experiments suggest that the real-time PCR assay is suitable for the detection of the ctxA gene in alkaline peptone water enrichments of potable water, surface water and sewage effluent samples.
REFERENCES
HUANG J, ZHU Y, WEN H, ZHANG J, HUANG S, NIU J and LI Q (2009) Quadruplex real time PCR assay for detection and identification of Vibrio cholerae O1 and O139 strains and determination of their toxigenic potential. Appl. Environ. Microbiol. 75 (22) 6981-6985. [ Links ]
KAPER JB, MORRIS JG and LEVINE MM (1995) Cholera. Clin. Microbiol. Rev. 8 (1) 48-86. [ Links ] KOSKELA KA, MATERO P, BLATNY JM, FYKSE EM, OLSEN JS,
NUOTIO LO and NIKKARI S (2009) A multiplatform real-time polymerase chain reaction detection assay for Vibrio cholerae. Diagn. Microbiol. Infect. Dis. 65 339-344.
MEHRABADI JF, MORSALI P, NEJAD HR and FOOLADI AAI (2012) Detection of toxigenic Vibrio cholerae with new multiplex PCR. J. Infect. Public Health 5 263-267. [ Links ]
MURPHY NM, McLAUCHLIN J, OHAI C and GRANT KA (2007) Construction and evaluation of a microbiological positive process internal control for PCR-based examination of food samples for Listeria monocytogenes and Salmonella enterica. Int. J. Food Microbiol. 120(1-2) 110-119. [ Links ]
ZHOU L, ZHANG K and WANNER BL (2004) Chromosomal expression of foreign and native genes from regulatable promoters in Escherichia coli. Methods Mol. Biol. 267123-134. [ Links ]
This paper was originally presented at the 2012 Water Institute of Southern Africa (WISA) Biennial Conference, Cape Town, 6-10 May 2012
* To whom all correspondence should be addressed +27 16 430-8403; fax: +27 16 455-2055 e-mail: nleat@randwater.co.za


{kind=link}
{kind=link}
{kind=link}