










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkWater SA
versión On-line ISSN 1816-7950
versión impresa ISSN 0378-4738
Water SA vol.39 no.3 Pretoria 2013
The impact of physico-chemical water quality parameters on bacterial diversity in the Vaal River, South Africa
Karen Jordaan; Cornelius Carlos Bezuidenhout*
School of Biological Sciences, Subject Group: Microbiology, North-West University, Potchefstroom Campus, Private Bag X6001, Potchefstroom 2520, South Africa
ABSTRACT
This study aimed to identify bacterial community structures in the Vaal River using PCR-DGGE (polymerase chain reaction denaturing gradient gel electrophoresis) and high-throughput sequencing. The impact of physico-chemical characteristics on bacterial structures was investigated through multivariate analysis. Samples were collected from 4 sampling stations along the Upper Vaal River during winter (June 2009) and summer (December 2010). Physico-chemical analysis was conducted on-site. Additional physico-chemical data were obtained from statutory bodies. DNA was directly isolated from water samples and PCR amplified using universal bacterial primer pairs. PCR products were subjected to DGGE fingerprinting and high-throughput sequencing, followed by Shannon-Weaver diversity calculations, cluster analysis and multivariate analysis. Physico-chemical parameters did not exceed the prescribed South African water quality standards for domestic use, aquatic ecosystems, livestock watering and irrigation. DGGE banding patterns revealed similar bacterial community structures for 3 of the 4 sampling stations. PCA and RDA indicated that pH, water temperature and inorganic nutrient concentrations could be used to explain changes in bacterial community structures. High-throughput sequencing data showed that bacterial assemblages were dominated by common freshwater groups: Cyanobacteria, Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, Bacteroidetes and Actinobacteria. Other freshwater phyla such as Deltaproteobacteria, Epsilonbacteria, Acidobacteria, Verrucomicrobia, Firmicutes, Fusobacteria, Flavobacteria and Fibrobacteres were found in low proportions. This study provides an overview of the dominant bacterial groups in the Upper Vaal River and the impact of environmental changes on bacterial diversity.
Keywords: Vaal River, bacterial community structures, 16S rDNA PCR-DGGE, high-throughput sequencing, multivariate analysis
INTRODUCTION
Socio-economic growth and development of the Vaal River require continuous augmentation of this water resource to meet the growing water requirements of communities in Gauteng, the Free State, North West and Northern Cape provinces (DWAF, 2009b). Water quality has drastically deteriorated due to constant disposal of industrial and domestic waste into the river. Salinisation, eutrophication and microbiological pollution are currently the main problems affecting the water quality (DWAF, 2009a). The Department of Water Affairs and Forestry (DWAF) of South Africa, in line with the South African National Water Act (NWA), Act No. 36 of 1998, stipulated regulatory guidelines and criteria a water system must meet to ensure that the country's water resources are fit for use. A structured biomonitoring programme was implemented by the DWAF in 2009 to determine the exact sensitivity and health status of the Vaal River (DWAF, 2009a). Criteria routinely monitored to ensure sustainability, optimal water use and protection of the water resource include: physico-chemical characteristics, stream flow, discharge loads and microbiological pollutants, in particular, Escherichia coli (DWAF, 2009a; 2009b). The detection of Escherichia coli only indicates the presence of faecal contamination and not necessarily the degree of industrial pollution. Therefore, in-depth studies on the microbial communities in the Vaal River are essential to understand the microbial processes underlying secondary pollution and changes in the physico-chemical quality of water.
DGGE has been applied in numerous research studies involving the assessment of microbial diversity of rivers, streams, lakes and sediment, to determine the water quality of the resource (De Figueiredo et al., 2010; Essahale et al., 2010; De Figueiredo et al., 2011; Haller et al., 2011). This method opened up new avenues of research on the diversity of microorganisms present in complex aquatic environments. Currently, metagenomic analysis of microbial ecology, such as high-throughput sequencing (HTS), has been the focus of several environmental studies such as soil, (Lemos et al., 2011), freshwater lakes (Marshall et al., 2008) and deep sea microbiota (Sogin et al., 2006). Metagenomic analysis provides extensive information on community structure and composition (Kakirde et al., 2010). In addition, phylogenetic and functional analyses of microorganisms can be determined at community level (Cowan et al., 2005).
The objectives of this study were (i) to identify the bacterial community structures in the planktonic phase of the Vaal River using 16S rDNA PCR-DGGE and high-throughput sequencing, and (ii) determine the impact of physico-chemical characteristics on bacterial community structures using principle component analysis (PCA) and redundancy analysis (RDA).
EXPERIMENTAL
Sample collection and physico-chemical analysis
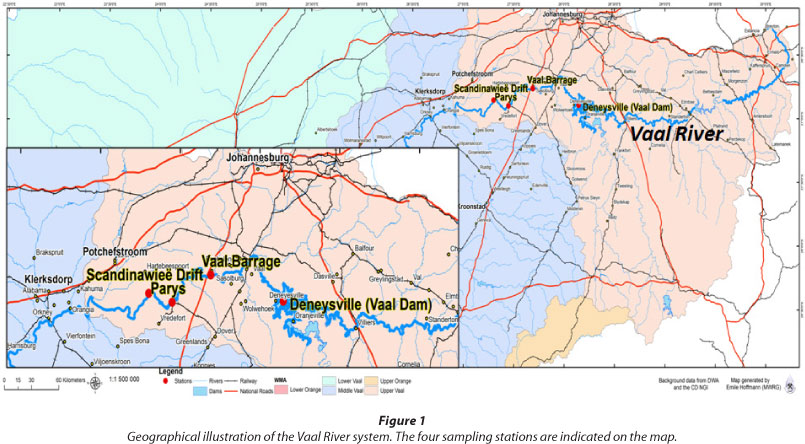
Water samples were collected from the Vaal River in June 2009 (winter) and December 2010 (summer). The four sites included Deneysville (Vaal Dam) (26°53'43.44"S 28°5'53.88"E), Vaal Barrage (26°45'53"S 27°41'30"E), Parys (26°54'0.36"S 27°26'60"E), and Scandinawieë Drift (26°51'20.45"S 27°18'9.52"E) (Fig. 1). The Vaal Dam and entire middle section of the Vaal River are respectively regarded as eutrophic and hypertrophic due to the high levels of chlorophyll-a and phosphate, which exceed the recommended standards (DWAF, 2009a).
Samples were collected from the planktonic phase in sterile glass bottles and preserved on ice for not longer than 6 h prior to nucleic acid isolation. Physico-chemical analysis was conducted in situ. Additional physico-chemical data were obtained from the Department of Water Affairs (2012) and the South African Weather Service (2012). A summary of the physico-chemical variables of all studied sampling sites is shown in Table 1.
Nucleic acid isolation
A hundred millilitres of water samples were filtered through a 0.45 µm nitrate cellulose membrane filter (Whatman, Missouri, USA) and subsequently lysed in a 1 mg/mℓ lysozyme solution that contained 0.25-0.50 mm glass beads (Sigma-Aldrich Co., Missouri, USA) for bacterial cell disruption. The lysis solution was incubated at 37°C for 10 min while agitated in a vortex. Proteinase K (1 mg/mℓ) was then added and the lysis solution was incubated at 56°C for an additional 30 min. DNA was isolated from the crude lysate using the PeqGold Bacterial DNA Kit (PEQLAB Biotechnologie GmbH, Erlangen, Germany). The quality and quantity of the isolated nucleic acids were determined using the Nanodrop ND1000 (NanoDrop Technologies, Delaware, USA) and agarose electrophoresis.
PCR amplification and DGGE analysis of bacterial community structures
The highly variable V3 region of the 16S rDNA gene fragments were PCR amplified using the universal primer pair 341F-GC and 907R (~ 500 bp) (Muyzer et al., 1993). Amplification was performed in 25 /µℓ reaction volumes containing single-strength PCR master mix ((5 U/µℓ Taq DNA polymerase (recombinant) in reaction buffer, 2 mM MgCl2, 0.2 mM of each dNTP, Fermentas Life Sciences, Maryland, USA)), 50 pmol of forward and reverse primers, additional 1 mM MgCl2, additional 1 Unit Taq DNA polymerase, 10-50 ng DNA and PCR-grade water (Fermentas Life Sciences, Maryland, USA). Thermal cycling was carried out in a Bio-Rad iCycler Thermal Cycler (Bio-Rad Laboratories, Hercules, California, USA) with an initial denaturation at 95°C for 7 min followed by 30 cycles of denaturation at 95°C for 30 s, annealing at 56°C for 1 min and extension at 72°C for 60 s. Final extension was performed at 72°C for 7 min. PCR products were evaluated by electrophoresis on 1% agarose gels and visualised by ethidium bromide staining and UV illumination.
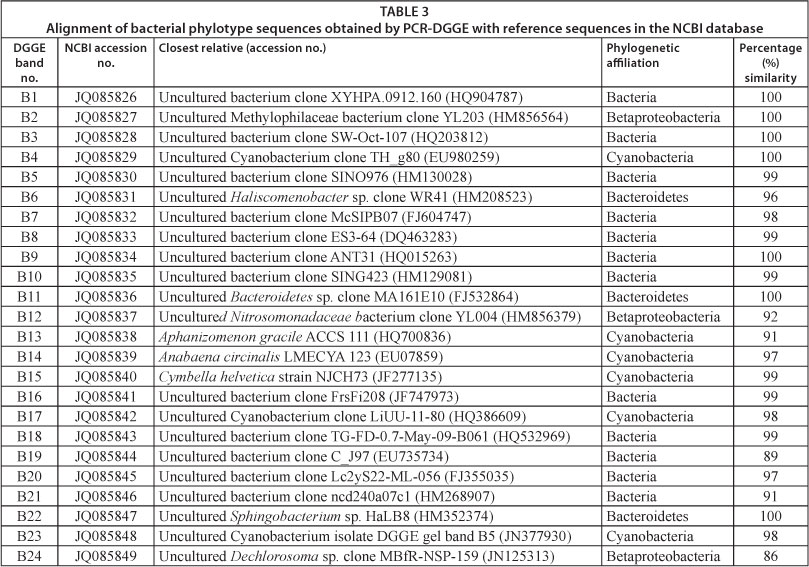
PCR products were analysed by DGGE using a DCode Universal Detection System (Bio-Rad Laboratories, Hercules, California, USA). Four reference species, namely Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus and Streptococcus faecalis, were included in all DGGE studies. DGGE analysis was conducted at a denaturing gradient of 30-50% in 1 mm vertical polyacrylamide gels (8% (wt/vol) acrylamide in 1 x TAE). 20 µℓ of amplification product were mixed with 5 µℓ of loading buffer (6χ Orange Loading Dye, Fermentas Life Sciences, Maryland, USA) and loaded into the gel. Electrophoresis was performed at a constant temperature of 60°C for 16 h at 100 V in 1 x TAE buffer (40 mM Tris-acetate, 1 mM EDTA, pH 8.0). Polyacrylamide gels were stained with ethidium bromide (10 mg/ℓ) for 45 min and visualised with a Gene Genius Bio Imaging System (Syngene, Cambridge, UK) and GeneSnap software (version 6.00.22). None of the DGGE gels were digitally enhanced or modified. Bands of interest were only highlighted for better visualisation and not analytical purposes. Selected DNA bands of interest were excised from gels with a sterile scalpel and eluted in 20 µℓ of sterile nuclease-free water for 12 h at 4°C. 2 µℓ of the elute were used as DNA template in PCR amplification reactions with primer pair 341F and 907R (Muyzer et al., 1993) and conditions described above. PCR products were subsequently purified and sequenced using a BigDye* Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, California, USA) and Genetic Analyzer 3130 (Applied Biosystems, California, USA). Sequences were aligned to 16S rRNA sequences in the National Center of Biotechnology Information Database (NCBI) using BLASTN searches to determine their identity. A total of 23 bacterial nucleotide sequences were submitted to the GenBank database under accession numbers JQ085826 - JQ085849.
High-throughput sequencing
HTS analysis was performed by Inqaba Biotech, South Africa, using the Roche 454 GS-FLXTM System. The V1-V3 region of the 16S rRNA gene was amplified using primer pair 27F (GAGTTTGATCCTGGCTCAG) and 518R (ATTACCGCGGCTGCTGG) to produce ~ 500 bp fragments. Subsequently, sequences were trimmed to remove GS tags and further analysed with the CLC Bio Genomics Workbench version 4.7.2 software (CLC Bio, Aarhus, Denmark). Sequences shorter than 200 bp in length were excluded from data sets. All remaining sequences were subjected to the National Center for Biotechnology Information (NCBI) database for BLAST analysis. Sequences were then submitted to Pintail version 1.0 to detect the presence of PCR artefacts. PCR products with chi-meric properties were eliminated from data sets prior to phylo-genetic analysis. The remaining 922 sequences were submitted to GenBank with accession numbers JN865256-JN866178.
Statistical analysis
Bacterial community diversity was calculated with the Shannon-Weaver diversity index (H'), based on DGGE profiles. The Shannon-Weaver indices (H') were calculated according to Zhang et al. (2011). Similarities between the banding patterns generated by PCR-DGGE of the various sampling sites were compared by cluster analysis as indicated by Gafan et al. (2005). Cluster analyses were displayed graphically as UPGMA dendrograms.
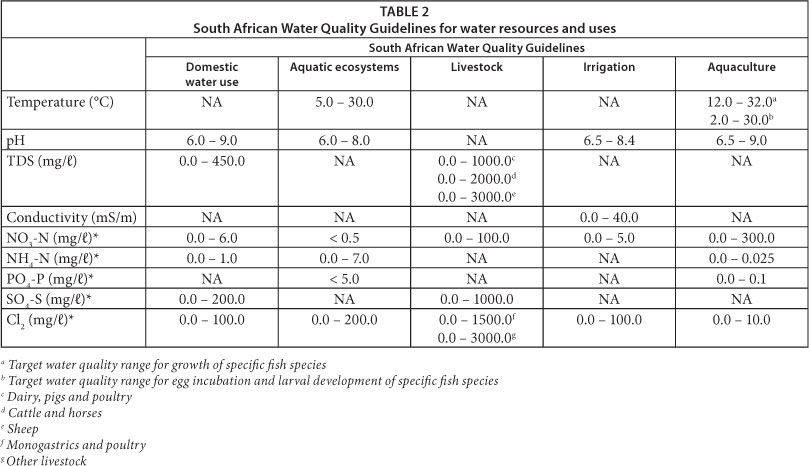
The distribution of samples according to environmental factors was analysed by PCA. The statistical significance of the relationships between bacterial community structures, DGGE banding profiles, high-throughput sequencing data and water quality was further assessed by RDA. Environmental variables selected are summarised in Table 2. Multivariate analysis was performed by a Monte Carlo permutations test using unlimited permutations. Analysis was carried out using CANOCO software version 4.5.
RESULTS
Physico-chemical characteristics
Selected physico-chemical parameters measured or obtained are listed in Table 1. These parameters showed all physico-chemical values to fall within the prescribed South African water quality guidelines for domestic use (DWAF, 1996a), aquatic ecosystems (DWAF, 1996b), livestock watering (DWAF, 1996c), irrigation (DWAF, 1996d) and aquaculture (DWAF, 1996e) (Table 2). Water temperatures were between 10 and 13°C in June and December temperatures exceeded 20°C (24.4-28.7°C). The temperatures of inland aquatic ecosystems in South Africa generally range between 5 and 30°C but can fluctuate depending on the geographical features of the region and catchment area, seasonal changes and the impact of anthropogenic activities (DWAF, 1996b). In December, the flow velocity increased sequentially from Deneysville to downstream sampling stations (Scandinawieë Drift). This trend was not observed in June, when rainfall was low.
Nucleic acid isolation from water samples
Nucleic acids were directly isolated from water samples without prior enrichment or culturing steps. Intact genomic DNA was obtained with a yield that varied from 2-30 ng/µℓ per 100 mℓ of water. The quality (A260:A280 ratio) of nucleic acids was acceptable for PCR and ranged from 1.6-2.2. Although DNA concentrations were low, amplification products were of sufficient quantity for PCR-DGGE analysis.
Dynamics of bacterial community structures
DGGE analysis
In this study, PCR-DGGE was able to give spatial information about the dominant bacterial communities in the Vaal River system (Fig. 2). Previous studies suggest that band intensity is related to the relative abundance of the corresponding phy-lotypes in the sample mixture (Murray et al., 1996; Riemann et al., 1999). Thus, bands with relatively high intensities were assumed to be dominant taxa.

DGGE profiles demonstrated high resolution and intensity at a denaturing gradient of 30-50%. Four bacterial species, Escherichia coli, Pseudomonas aeruginosa, Streptococcus faecalis and Staphylococcus aureus, were included in all DGGE studies, to determine the potential of using such an approach to establish the presence of these species in water samples. Corresponding bands for Staphylococcus aureus and Pseudomonas aeruginosa were detected for Vaal Barrage, Parys and Scandinawieë Drift. In addition, Parys illustrated a band with similar migration patterns to Escherichia coli. All corresponding bands were excised and sequenced but produced poor quality sequences with indefinite identification. Since sequence data could not confirm accurate identification of excised bands, results remain inconclusive.
Vaal Barrage, Parys and Scandinawieë Drift displayed similar DGGE patterns for the dominant bands in June and December (Fig. 2). However, DGGE profiles for Deneysville varied to some extent from the three other sites. Although some dominant bands showed similar migration patterns to Vaal Barrage, Parys and Scandinawieë Drift, a few distinct bands exhibited unique migration positions. A higher bacterial diversity, based on number of bands, was detected for Vaal Barrage and Scandinawieë Drift during June compared to December. On the other hand, bacterial diversity for Deneysville was higher in December than in June. The Shannon-Weaver indices (Fig. 4), however, contradicted the DGGE diversity data. These showed a higher bacterial diversity for Vaal Barrage and Scandinawieë Drift during December compared to June. The Shannon-Weaver index calculation includes the presence and absence of bands, but also band intensity that could be used to explain the contradiction (Zhang et al., 2011).

A total of 24 bacterial bands were excised, sequenced and compared to sequences in the NCBI database (Table 3). Approximately 75% of the bacterial sequences recovered displayed high sequence homologies (> 97%) with the known database sequences. However, 50% of these sequences showed the highest sequence similarity to uncultured bacteria obtained directly from freshwater samples. These results support the presence of many uncultured and potentially undescribed bacterial taxa in freshwater ecosystems. Taxonomic classifications of the partial 16S rDNA sequences obtained affiliated to Cyanobacteria (B4, B13-B15, B17, B23), Bacteroidetes (B6, B11, B22), Betaproteobacteria (B2, B12, B24) and uncultured bacteria (B1, B3, B5, B7-B10, B16, B18-B21). Bacterial communities for June displayed relative abundances of 8%, 17%, 17% and 58% for Cyanobacteria, Bacteroidetes, Betaproteobacteria and uncultured bacteria, respectively. In contrast, the relative abundance for Cyanobacteria increased to 42% in December, whereas Bacteroidetes, Betaproteobacteria, and uncultured bacteria respectively accounted for 8%, 8% and 42% of the four main phylogenetic groups.
High-throughput sequencing
A total of 18 phyla were identified among 4 sampling sites by HTS technology (Fig. 3A - F). Dominant phyla include Alphaproteobacteria (0.24-15%), Betaproteobacteria (1.4785.10%), Gammaproteobacteria (0.24-12.38%), Bacteroidetes (0.72-4.05%) and Actinobacteria (4.76-10.00%). The remaining groups could be placed into 9 phyla: Acidobacteria, Chloroflexi, Cyanobacteria, Euglenoidea, Eukaryote, Fibrobacteres, Firmi-cutes, Fusobacteria, and Verrucomicrobia.
While identification of the four indicator organisms employed in DGGE profiling remained inconclusive by Sanger sequencing, HTS analysis verified that two of the bands did in fact belong to the Pseudomonadaceae family and Escherichia spp. Additional opportunistic pathogens detected in low quantities at Vaal Barrage, Parys and Scandinawieë Drift included Roseomonas sp., Ralstonia sp., Serratia sp. and Stenotrophomonas sp.
Distribution of bacterial diversity in the Vaal River
The Shannon-Weaver diversity indices (H') were calculated from DGGE banding patterns as the number and relative intensity of bands (Fig. 4). Indices were used to compare the overall structure of bacterial communities among the four sampling sites. H' for June and December samples ranged from 0.27-0.46 and 0.70-0.86, respectively. Bacterial diversity gradually increased from upstream to downstream sites, except for Parys in December which consisted of a lower diversity. Similar trends were also observed for HTS data.
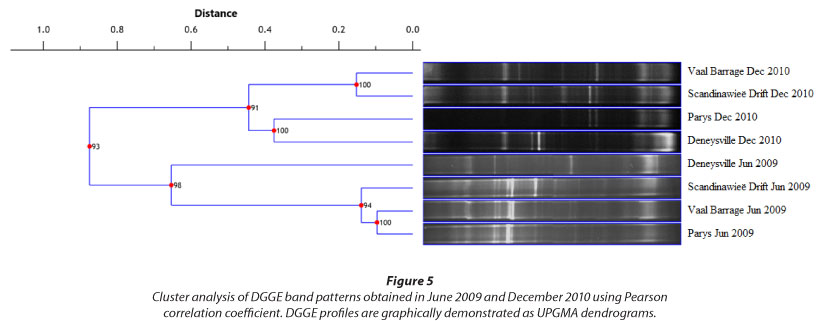
Cluster analysis was performed to gain an overview of the association of bacterial communities at the four sampling stations during June and December (Fig. 5). UPMGA dendrograms showed grouping of samples according to season. June samples showed high similarity (> 94%) among bacterial communities for Vaal Barrage, Parys and Scandinawieë Drift. A similar trend was observed for the December samples where Vaal Barrage and Scandinawieë Drift were defined by a 100% similarity. Noticeable was the grouping of the December Parys and Deneysville samples (100% similarity). Grouping of these two sampling sites may be attributed to similar banding patterns of a few dominant DGGE bands (Fig. 2). Diversity indices (H') and cluster analyses could be associated with DGGE profiles which reflected variations in the distribution, abundance and composition of bacterial taxa.
Multivariate analysis
PCA and RDA were performed to analyse the relationships between the environmental parameters and the clustering of samples.
The effect of different sampling periods is illustrated by the PCA analysis results (Fig. 6A). The June samples, with negativeb and positive scores along the first axis, are separated from the December samples, which showed a positive score along the second axis. The first axis was mainly defined by ammonium, nitrate, phosphate, chloride, sulphate, TDS, conductivity and rainfall. The second axis was related to temperature, day length and flow rate.
RDA plots calculated from DGGE profiles highlighted the possible environmental parameters responsible for the distribution of bacterial species at the different sampling stations (Figs. 6B and C). The arrow vectors for the environmental parameters in each RDA plot represent their impact on the composition of bacterial communities. Variation in the distribution of bacterial communities for the June and December samples (Figs. 6B and C) was shown to be correlated with the pH (BN8, BN14), temperature (BN11, BN15), ammonium (BN9, BN4, BN18, BN19 and BN22), phosphate (BN9, BN4, BN16 and BN17), chloride (BN3, BN5, BN16 and BN17), sulphate (BN3, BN5, BN18, BN19 and BN22), nitrate (BN11) and TDS concentrations (BN3, BN5, BN16 and BN17).
RDA plots for high-throughput sequencing data (Fig. 6D) showed (i) positive correlations between the flow rate and abundances of Gammaproteobacteria, Deltaproteobacteria and Fibrobacteres along the first axis, (ii) positive correlations between rainfall, TDS, nitrate, ammonium, chloride and sulphate concentrations, and abundances of Acidobacteria and Actinobacteria along the second axis, and (iii) positive correlations between ammonium, chloride and phosphate concentrations, and abundances of Fusobacteria, Verrucomicrobia and Euglenoida along the second axis. Betaproteobacteria negatively correlated with Gammaproteobacteria. A high abundance of Betaproteobacteria was detected in June but decreased considerably in December. An opposite inclination was observed for Gammaproteobacteria.
DISCUSSION
Microbial community dynamics
Knowledge and insight into the diversity and function of freshwater microorganisms is an essential requirement for the sustainable management of freshwater resources. In addition, changes in bacterial community structures might be used as potential bio-indicators of environmental disturbances. The aim of this study was to examine bacterial community structures in a segment of the Vaal River, in response to environmental parameters, using a PCR-DGGE and high-throughput sequencing approach. High-throughput sequencing provided an overview of the dominant bacterial communities in the planktonic phase and marked shifts in composition, as attested to by PCA and RDA.
The composition of bacterial communities in a given environment depends on the interaction between various factors such as the geographic environment (Zhang et al., 2011), temperature (Hall et al., 2008), pH (Yannarell and Triplett, 2005), flow rate (Crump and Hobbie, 2005), light intensity (Sigee, 2005) and nutrient concentrations (Pomeroy and Wiebe, 2001). In this study of a segment of the Vaal River, the physico-chemical parameters varied with sampling station and season of sampling. PCA and RDA analysis indicated that bacterial community structures were mainly influenced by pH, temperature and inorganic components.
The bacterial community structures were similar for the three sampling sites during each sampling period. However, the June bacterial community structures were different to the December assemblages. DGGE results suggested that bacterial diversity was higher during June compared to December. These results were, however, contradicted by the Shannon-Weaver indices. The latter analysis included presence-absence, as well as (abundance) band intensity data. This could be used to explain the contradiction (Zhang et al., 2011). Diversity index analysis of the high-throughput sequencing data showed similar trends to the Shannon-Weaver analysis of DGGE profiles.
Bacterial community structures could be correlated to inorganic nutrients as shown by PCA and RDA. The Vaal Barrage creates a buffering action that encapsulates organic and inorganic particles in the water-column for several weeks. This creates a relatively stable environment in which organisms can develop into a community. The planktonic bacteria then flow from here downstream to Parys and Scandinawieë Drift. Therefore, bacterial communities along this section of the Vaal River will be relatively similar. In addition, the dominant bacterial groups detected at these three points may be native species with broader niche capabilities, which allow them to grow and survive under a variety of environmental conditions (Anderson-Glenna et al., 2008). Recurrent native bacterial communities in aquatic ecosystems have been reported previously (Sekiguchi et al., 2002; Crump et al., 2003). It should be noted that the DNA amplification method used in this study did not discriminate between DNA derived from living cells versus DNA from dead cells and/or even naked or free DNA available in the water column. This aspect should be considered in future aquatic studies.
A feature highlighted in the present study was the relatively low bacterial diversity detected at Deneysville in June and December. Bacterial community structures at this sampling station largely consisted of Cyanobacteria, particularly Cyanophyta (Anabaena sp.), where pH and temperature were the main factors that affected the community structures. An alkaline pH was measured in June and December when temperatures in December were above 25°C. Optimum growth of Cyanophyta and the formation of surface algal blooms are the direct result of high nutrient concentrations (particularly phosphate) and physico-chemical characteristics (high pH, temperature and light intensity) (Sigee, 2005). In addition to these conditions, buoyancy also plays an important role in the development of Cyanophyta populations. Buoyancy allows algal populations to adopt an optimum position within the water column in relation to light and CO2 availability (Sigee, 2005). This mechanism leads to changes in the water chemistry and light regime in the epilimnion that depress the growth of other phyto- and bacterioplankton groups (Sigee, 2005).
Although flow rate in this study was not shown to affect bacterial communities, previous studies have suggested that flow rate and hydraulic retention time have a substantial effect on community structures (Lindström and Bergström, 2004; Crump and Hobbie, 2005). Temporal variation in bacterial diversity was observed between the June and December samples. The Gauteng and North West Provinces received heavy rainfall in December 2010 that caused a drastic increase in flow rate, particularly at Parys and Scandinawieë Drift. The high flow rate resulted in flooding at these two sampling stations that likely changed the bacterial community structures. Bacterial communities in rivers with short hydraulic retention times would potentially remain undetected by DGGE due to high loss rates (wash-out effect) which in turn result in a lower bacterial density and diversity (Sommaruga and Casamayor, 2009). In contrast, rivers with an extended hydraulic retention time display an accumulation of nutrients which promotes a higher genetic diversity of bacteria. Although flow rate differences provide a reasonable explanation for the seasonal variation in bacterial, further investigations are needed to confirm this for the Vaal River.
Phylogenetic diversity of bacterial communities
Phylogenetic affiliation of the dominant groups retrieved from the freshwater samples by PCR-DGGE and high-throughput sequencing corresponded to Cyanobacteria, Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, Bacteroidetes and Actinobacteria. Other freshwater phyla such as Deltaproteobacteria, Epsilonbacteria, Acidobacteria, Verrucomicrobia, Firmicutes, Fusobacteria, Flavobacteria and Fibrobacteres were found in low proportions.
Cyanobacteria accounted for a large proportion of bacterial diversity during December, which agrees well with the physico-chemical characteristics of the water samples. Several studies have indicated that Cyanobacteria tend to dominate phytoplankton communities in pristine freshwater systems (Anderson-Glenna et al., 2008; Foong et al., 2010) whereas other authors have reported an increase in the prevalence of Cyanobacteria in response to fluvial, organic and urban wastewater pollution (Douterelo et al., 2004; Ibekwe et al., 2012). Due to the trophic status of the Vaal River, cyanobac-terial blooms usually occur during late spring and summer and often consist of Microcystis aeruginosa, Oscillatoria sp. and Anabaena floss-aqua (Cloot and Le Roux, 1997; DWAF, 2009a). In this study, Anabaena sp., Cymbella helvetica and Synechocystis sp. were in high abundance at Deneysville during December 2010. Anabaena spp. are among the most distributed toxin producers in eutrophic freshwater bodies (Berg et al., 1986). Their potential effects on aquatic ecosystems may be subtle or can cause major changes in the survival of sensitive species (DWAF, 2009a). In addition, these toxins may pose a serious health hazard for human and animal consumption.
Alphaproteobacteria, Betaproteobacteria, Gammaproteo-bacteria and Actinobacteria are ubiquitous groups in freshwater habitats (Gich et al., 2005; Anderson-Glenna et al., 2008) and are numerically important in river systems (Beier et al., 2008; Lemke et al., 2009). Members of Betaproteobacteria respond rapidly to organic and inorganic nutrient enrichment (Hahn, 2003; Simek et al., 2005) and have been isolated from various polluted and unpolluted freshwater bodies (De Figueiredo et al., 2011; Haller et al., 2011). Two important genera of this subphylum include Dechlorosomonas and Variovorax. Members of Dechlorosomonas are capable of oxidising aromatic compounds such as benzoate, chlorobenzo-ate and toluene (Coates et al., 2001), whereas Variovorax spp. are involved in plant growth and remediation of xenobiotics (Jamieson et al., 2009). Several opportunistic human pathogens of the Gammaproteobacteria group were detected at low abundance. Human diseases and infections are often associated with these pathogens (Berg et al., 2005; Mahlen, 2011) and have caused mortalities in immunocompromised individuals (Fergie et al., 1994; Paez and Costa 2008). Thus, although the opportunistic pathogens were present at low levels, their impact should not be underestimated.
RDA analysis revealed that nitrate, ammonium, chloride and sulphate were the four most influential inorganic factors responsible for shaping Actinobacterial and Acidobacterial communities. A few studies suggested that these two phyla participate in the nitrogen cycle in soils and sediments by reducing nitrate, nitrite and possibly nitric oxide (Gtari et al., 2007; Ward et al., 2009). Norris et al. (2011) also implicated some novel Actinobacteria from geothermal environments in growing autotrophically with sulphur as an energy source. Correlation between Verrucomicrobia and phosphate was also detected suggesting that this inorganic nutrient influenced the Verrucomicrobia community within the total bacterial population. The association between Verrucomicrobia and phosphate levels has seldom been discussed in previous studies of microbial ecology of freshwater resources (Lindström et al., 2005; Liu et al., 2009). Very little is known about the physiology and ecological roles of Actinobacteria, Acidobacteria and Verrucomicrobia in these habitats and the impact of physico-chemical characteristics on their community composition.
Members of Bacteroidetes usually inhabit mesotrophic and eutrophic water bodies that have high nutrient levels (Xi et al., 2007; de Figueiredo et al., 2011). This group is known to degrade polymeric organic matter, and to play an important role in the turnover of organic matter (Cottrell and Kirchman, 2000), and is often isolated from humic waters (Anderson-Glenna et al., 2008; Stabili and Cavallo, 2011). The Bacteroidetes-Flavobacterium-like lineages are often present in high abundance following the growth and decline of cyano-bacterial blooms (Eiler and Bertilsson, 2007; Newton et al., 2011). Their presence and distribution is mainly determined by resource availability and is favoured during periods of high het-erotrophic activity and enhanced growth (Eiler and Bertilsson, 2007). This phenomenon was evident in the high abundance of Bacteroidetes in June following the December 2008 to February 2009 cyanobacterial blooms.
CONCLUSIONS
This study investigated the impact of physico-chemical water quality parameters on bacterial community structures in a segment of the Vaal River. The PCR-DGGE approach and high-throughput sequencing analysis presented useful data in the identification of dominant bacterial groups at the four sampling stations. Molecular analysis showed that (i) bacterial community structures for June were different to the December assemblages, (ii) bacterial community structures for Vaal Barrage, Parys and Scandinawieë Drift were similar, (iii) bacterial communities at Deneysville differed from the three other sites and were lower in diversity, and (iv) Cyanobacteria, Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, Bacteroidetes and Actinobacteria were the dominant bacterial groups detected and were shown to be impacted by physico-chemical water quality parameters. This study contributed to the identification of bacterial phylotypes, their spatial succession and the effect of physico-chemical characteristics on these freshwater bacterial communities. A detailed study on the relationships between the dominant bacterial taxa and specific physico-chemical water characteristics is required to improve our knowledge on how bacterial community structures in the Vaal River are affected.
ACKNOWLEDGEMENTS
The authors wish to thank the Water Research Commission (WRC) and North-West University for financial contributions. This research forms an integral part of a WRC-funded project (K5/1966) on water quality in the North West Province.
REFERENCES
ANDERSON-GLENNA M J, BAKKESTEUN V and CLIPSON NJW (2008) Spatial and temporal variability in epilithic biofilm bacterial communities along an upland river gradient. FEMS Microbiol. Ecol. 64 407-418. [ Links ]
BEIER S, WITZEL K-P and MARXSEN J (2008) Bacterial community composition in Central European running waters examined by temperature gradient gel electrophoresis and sequence analysis of 16S rRNA genes. Appl. Environ. Microbiol. 74(1) 188-199. [ Links ]
BERG K, SKULBERG OM, SKULBERG R, UNDERDAL B and WILLEN T (1986) Observations of toxic blue-green algae (Cyanobacteria) in some Scandinavian lakes. Acta Vet. Scand. 27 440-452. [ Links ]
BERG G, EBERL L and HARTMANN A (2005) The rhizosphere as a reservoir for opportunistic human pathogenic bacteria. Environ. Microbiol. 7 (11) 1673-1685. [ Links ]
CLOOT A and LE ROUX G (1997) Modelling algal blooms in the Middle Vaal River: a site specific approach. Water Res. 31 (2) 271-279. [ Links ]
COATES JD, CHAKRABORTY R, LACK JG, O'CONNOR SM, COLE KA, BENDER KS and ACHENBACH LA (2001) Anaerobic benzene oxidation coupled to nitrate reduction in pure culture by two strains of Dechloromonas. Nature 4111039-1043. [ Links ]
COTTRELL MT and KIRCHMAN DL (2000) Natural assemblages of marine Proteobacteria and members of the Cytophaga-Flavobacter cluster consuming low- and high-molecular weight dissolved organic matter. Appl. Environ. Microbiol. 66 1692-1697. [ Links ]
COWAN D, MEYER Q, STAFFORD W, MUYANGA S, CAMERON R and WITTWER P (2005) Metagenomic gene discovery, past, present and future. Trends Biotechnol. 23321-329. [ Links ]
CRUMP BC, KLING GW, BAHR M and HOBBIE J (2003) Bacterioplankton community shifts in an Arctic lake correlate with seasonal changes in organic matter source. Appl. Environ. Microbiol. 69 2253-2268. [ Links ]
CRUMP BC and HOBBIE JE (2005) Synchrony and seasonality in bacterioplankton communities of two temperate rivers. Limnol. Oceanogr. 50 (6) 1718-1729. [ Links ]
DE FIGUEIREDO DR, PEREIRA MJ and CORREIA A (2010) Seasonal modulation of bacterioplankton community at a temperate eutrophic shallow lake. World J. Microbiol. Biotechnol. 261067-1077. [ Links ]
DE FIGUEIREDO DR, FERREIRA RV, CERQUEIRA M, CONDESSO DE MELO T, PEREIRA MJ, CASTRO BB and CORREIA A (2011) Impact of water quality on bacterioplankton assemblage along Cértima River Basin (central western Portugal) assessed by PCR-DGGE and multivariate analysis. Environ. Monit. Assess. 184 (1) 471-485. [ Links ]
DEPARTMENT OF WATER AFFAIRS (2012) URL: http://www.dwa.gov.za/ (Accessed 23 January 2012). [ Links ]
DOUTERELO I, PERONA E and MATEO P (2004) Use of cyanobacteria to assess water quality in running waters. Environ. Pollut. 127 377-384. [ Links ]
DWAF (DEPARTMENT OF WATER AFFAIRS AND FORESTRY, SOUTH AFRICA) (1996a) South African Water Quality Guidelines (2nd edn.) Volume 1: Domestic Use. Department of Water Affairs and Forestry, Pretoria. [ Links ]
DWAF (DEPARTMENT OF WATER AFFAIRS AND FORESTRY, SOUTH AFRICA) (1996b) South African Water Quality Guidelines. Volume 7: Aquatic Ecosystems. Department of Water Affairs and Forestry, Pretoria. [ Links ]
DWAF (DEPARTMENT OF WATER AFFAIRS AND FORESTRY, SOUTH AFRICA) (1996c) South African Water Quality Guidelines (2nd edn.) Volume 5: Agricultural Use: Livestock Watering. Department of Water Affairs and Forestry, Pretoria. [ Links ]
DWAF (DEPARTMENT OF WATER AFFAIRS AND FORESTRY, SOUTH AFRICA) (1996d) South African Water Quality Guidelines (2nd edn.) Volume 4: Agricultural Use: Irrigation. Department of Water Affairs and Forestry, Pretoria. [ Links ]
DWAF (DEPARTMENT OF WATER AFFAIRS AND FORESTRY, SOUTH AFRICA) (1996e) South African Water Quality Guidelines (2nd edn.) Volume 6: Agricultural Water Use: Aquaculture. Department of Water Affairs and Forestry, Pretoria. [ Links ]
DWAF (DEPARTMENT OF WATER AFFAIRS AND FORESTRY, SOUTH AFRICA) (2009a) Integrated water quality management plan for the Vaal River System: Task 2: Water quality status assessment of the Vaal River System. Report No. P RSA C000/00/2305/1. September 2009. Directorate: National Water Resource Planning. Department of Water Affairs and Forestry, Pretoria. [ Links ]
DWAF (DEPARTMENT OF WATER AFFAIRS AND FORESTRY, SOUTH AFRICA) (2009b) Vaal River System: Large Bulk Water Supply Reconciliation Strategy: Executive Summary. March 2009. Department of Water Affairs and Forestry, South Africa. [ Links ]
EILER A and BERTILSSON S (2007) Flavobacteria blooms in four eutrophic lakes: linking population dynamics of freshwater bacte-rioplankton to resource availability. Appl. Environ. Microbiol. 73 3511-3518. [ Links ]
ESSAHALE A, MALKI M, MARÍN I and MOUMNI M (2010) Bacterial diversity in Fez tanneries and Morocco's Binlamdoune River, using 16S RNA gene based fingerprinting. J. Environ. Sci. 22 (12) 1944-1953. [ Links ]
FERGIE JE, SHEMA SJ, LOTT L, CRAWFORD R and PATRICK CC (1994) Pseudomonas aeruginosa bacteremia in immunocompro-mised children: analysis of factors associated with a poor outcome. Clin. Infect. Dis. 18(3) 390-394. [ Links ]
FOONG CP, LING CMWV and GONZALEZ M (2010) Metagenomic analyses of the dominant bacterial community in the Fildes Peninsula, King George Island (South Shetland Islands). Polar Sci. 4 263-273. [ Links ]
GAFAN GP, LUCAS VS, ROBERTS GJ, PETRIE A, WILSON M and SPRATT DA (2005) Statistical analyses of complex denaturing gradient gel electrophoresis profiles. J. Clin. Microbiol. 43(8) 3972-3978. [ Links ]
GICH F, SCHUBERT K, BRUNS A, HOFFELNER H and OVER- MANN J (2005) Specific detection, isolation, and characterization of selected, previously uncultured members of the freshwater bacterioplankton community. Appl. Environ. Microbiol. 71 (10) 5908-5919. [ Links ]
GTARI M, BRUSETTI L, HASSEN A, MORA D, DAFFONCHIO D and BOUDABOUS A (2007) Genetic diversity among Elaeagnus compatible Frankia strains and sympatric-related nitrogen-fixing actinobacteria revealed by nifH sequence analysis. Soil Biol. Biochem. 39 372-377. [ Links ]
HAHN MW (2003) Isolation of strains belonging to the cosmopolitan Polynucleobacter necessarius cluster from freshwater habitats located in three climatic zones. Appl. Environ. Microbiol. 69 5248-5254. [ Links ]
HALL EK, NEUHAUSER C and COTNER JB (2008) Toward a mechanistic understanding of how natural bacterial communities respond to changes in temperature in aquatic ecosystems. ISME 2 471-481. [ Links ]
HALLER L, TONOLLA M, ZOPFI J, PEDUZZI R, WILDI W and POTÉ J (2011) Composition of bacterial and archaeal communities in freshwater sediments with different contamination levels (Lake Geneva, Switzerland). Water Res. 451213-1228. [ Links ]
IBEKWE AM, LEDDY MB, BOLD RM and GRAVES AK (2012) Bacterial community composition in low-flowing river water with different sources of pollutants. FEMS Microbiol. Ecol. 79155-166. [ Links ]
JAMIESON WD, PEHL MJ, GREGORY GA and ORWIN PM (2009) Coordinated surface activities in Variovorax paradoxus EPS. BMC Microbiol. 9 (124) 1-18. [ Links ]
KAKIRDE KS, PARSLEY LC and LILES MR (2010) Size does matter: Application-driven approaches for soil metagenomics. Soil Biol. Biochem. 42 4399-4406. [ Links ]
LEMKE MJ, LIENAU EK, ROTHE J, PAGIORO TA, ROSENFELD J and DESALLE R (2009) Description of freshwater bacterial assemblages from the Upper Parana River Floodpulse System, Brazil. Microbiol. Ecol. 5794-103. [ Links ]
LEMOS LN, FULTHORPE RR, TRIPLETT EW and ROESCH LFW (2011) Rethinking microbial diversity analysis in the high throughput sequencing era. J. Microbiol. Meth. 8642-51. [ Links ]
LINDSTRÖM ES and BERGSTRÖM A-K (2004) Influence of inlet bacteria on bacterioplankton assemblage composition in lakes of different hydraulic retention time. Limnol. Oceanogr. 49 125-136. [ Links ]
LINDSTRÖM ES, KAMST-VAN AGTERVELD MP and ZWART G (2005) Distribution of typical freshwater bacterial groups is associated with pH, temperature, and lake water retention time. Appl. Environ. Microbiol. 71(12) 8201-8206. [ Links ]
LIU FH, LIN GH, GAO G, QIN BQ, ZHANG JS, ZHAO GP, ZHOU ZH and SHEN JH (2009) Bacterial and archaeal assemblages in sediments of a large shallow freshwater lake, Lake Taihu, as revealed by denaturing gradient gel electrophoresis. J. Appl. Microbiol. 1061022-1032. [ Links ]
MAHLEN SD (2011) Serratia infections: from military experiments to current practice. Clin. Microbiol. Rev. 24(4) 755-791. [ Links ]
MARSHALL MM, AMOS RN, HENRICH VV and RUBLEE PA (2008) Developing SSU rDNA metagenomic profiles of aquatic microbial communities for environmental assessments. Ecol. Indic. 8 442-453. [ Links ]
MURRAY AE, HOLLIBAUGH JT and ORREGO C (1996) Phylogenetic compositions of bacterioplankton from two California estuaries compared by denaturing gradient gel electrophoresis of 16S rDNA fragments. Appl. Environ. Microbiol. 62 (7) 2676-2680. [ Links ]
MUYZER G, DE WAAL EC and UITTERLINDEN AG (1993) Profiling of complex microbial populations by encoding for 16S rRNA. Appl. Environ. Microbiol. 59695-700. [ Links ]
NEWTON RJ, JONES SE, EILER A, MCMAHON KD and BERTILS-SON S (2011) A guide to the natural history of freshwater lake bacteria. Microbiol. Mol. Biol. R. 75 (1) 14-49. [ Links ]
NORRIS PR, DAVIS-BELMAR CS, BROWN CF and CALVO-BADO LA (2011) Autotrophic, sulfur-oxidizing actinobacteria in acidic environments. Extremophiles 15(2) 155-163. [ Links ]
PAEZ JIG and COSTA SF (2008) Risk factors associated with mortality of infections caused by Stenotrophomonas maltophilia: a systematic review. J. Hosp. Infect. 70 101-108. [ Links ]
POMEROY LR and WIEBE WJ (2001) Temperature and substrates as interactive limiting factors for marine heterotrophic bacteria. Aquat. Microb. Ecol. 23 (2) 187-204. [ Links ]
RIEMANN L, STEWARD GF, FANDINO LB, CAMPBELL L, LANDRY MR and AZAM F (1999) Bacterial community composition during two consecutive NE monsoon periods in the Arabian Sea studied by denaturing gradient gel electrophoresis (DGGE) of rRNA genes. Deep Sea Res. 46(8-9) 1791-1811. [ Links ]
SEKIGUCHI H, WATANABE M, NAKAHARA T, XU B and UCHIYAMA H (2002) Succession of bacterial community structure along the Changjiang River determined by denaturing gradient gel electrophoresis and clone library analysis. Appl. Environ. Microbiol. 68 (10) 5142-5150. [ Links ]
SIGEE DC (2005) Freshwater Microbiology: Biodiversity and Dynamic Interactions of Microorganisms in the Aquatic Environment. John Wiley & Sons Ltd, West Sussex, England. [ Links ]
SIMEK K, HORNAK K, JEZBERA J, MASIN M, NEDOMA J, GASOL J and SCHAUER M (2005) Influence of top-down and bottom-up manipulations on the R-BT065 subcluster of β-Proteobacteria an abundant group in bacterioplankton of a freshwater reservior. Appl. Environ. Microbiol. 71 2381-2390. [ Links ]
SOGIN ML, MORRISON HG, HUBER JA, WELCH DM, HUSE SM, NEAL PR, ARRIETA JM and HERNDL GJ (2006) Microbial diversity in the deep sea and the underexplored "rare biosphere". P. Natl. Acad. Sci. USA 103 12115-12120. [ Links ]
SOMMARUGA R and CASAMAYOR EO (2009) Bacterial 'cosmopolitanism' and importance of local environmental factors for community composition in remote high-altitude lakes. Freshwater Biol.55 (5) 994-1005. [ Links ]
SOUTH AFRICAN WEATHER SERVICE (2012) URL: http://www.weathersa.co.za/web/ (Accessed 23 January 2012). [ Links ]
STABILI L and CAVALLO RA (2011) Microbial pollution indicators and culturable heterotrophic bacteria in a Mediterranean area (Southern Adriatic Sea Italian coasts). J. Sea Res. 65461-469. [ Links ]
WARD NL, CHALLACOMBE JF, JANSSEN PH, HENRISSAT B, COUTINHO PM, WU M, XIE G, HAFT DH, SAIT M, BADGER J, BARABOTE RD, BRADLEY B, BRETTIN TS, BRINKAC LM, BRUCE D, CREASY T, DAUGHERTY SC, DAVIDSEN TM, DEBOY RT, DETTER JC, DODSON RJ, DURKIN AS, GANAPATHY A, GWINN- GIGLIO M, HAN CS, KHOURI H, KISS H, KOTHARI SP, MADUPU R, NELSON KE, NELSON WC, PAULSEN I, PENN K, REN Q, ROSOVITZ MJ, SELENGUT JD, SHRIVASTAVA S, SULLIVAN SA, TAPIA R, THOMPSON LS, WATKINS KL, YANG Q, YU C, ZAFAR N, ZHOU L and KUSKE CR (2009) Three genomes from the phylum Acidobacteria provide Insight into the lifestyles of these microorganisms in soils. Appl. Environ. Microbiol. 75(7) 2046-2056. [ Links ]
XI W, WU X, YE W and YANG H (2007) Changes in bacterial community structure during preceding and degraded period of cyano-bacterial bloom in a bay of the Taihu Lake. Chin. J. Appl. Environ.Biol. 13(1) 97-103. [ Links ]
YANNARELL AC and TRIPLETT EW (2005) Geographic and environmental sources of variation in lake bacterial community composition. Appl. Environ. Microbiol. 71 (1) 227-239. [ Links ]
ZHANG S, YANG G, HOU S and WANG Y (2011) Abundance and diversity of glacial bacteria on the Tibetan Plateau with environment. Geomicrobiol. J. 27(8) 649-655. [ Links ]
This paper was originally presented at the 2012 Water Institute of Southern Africa (WISA) Biennial Conference, Cape Town, 6-10 May 2012.
* To whom all correspondence should be addressed. +27 18 299-2315; fax: +27 18 299-2330; -mail: carlos.bezuidenhout@nwu.ac.za


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}